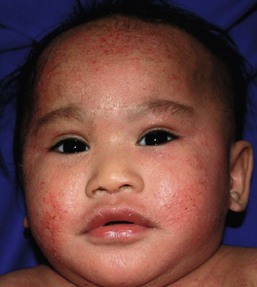
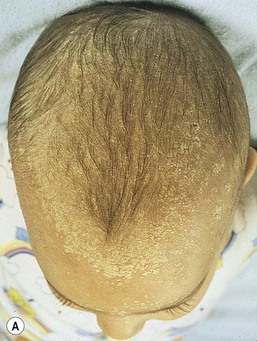
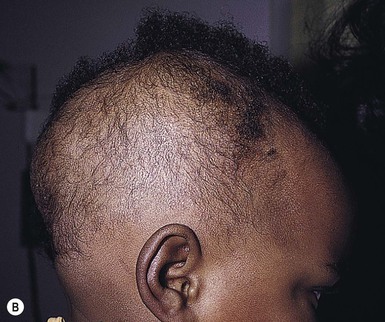
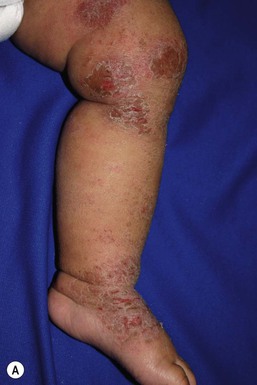
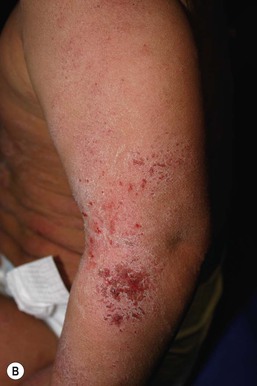
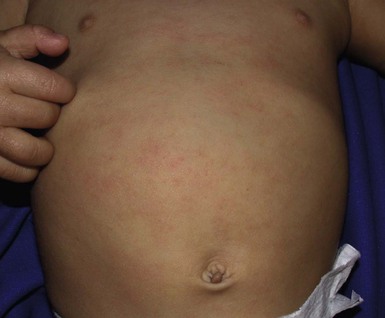
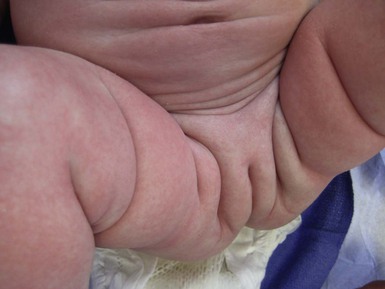
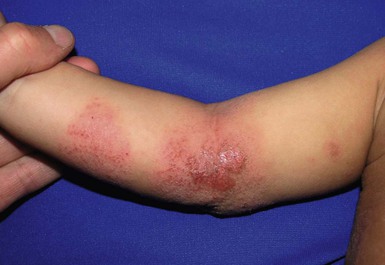
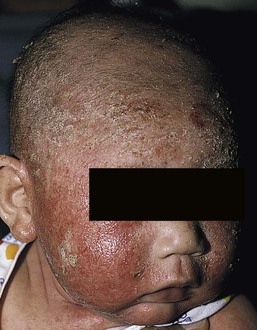
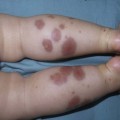
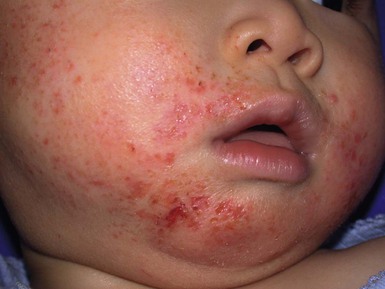
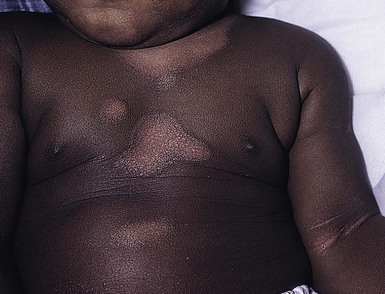
Wynnis L. Tom, lawrence F. Eichenfield Eczematous eruptions represent a significant proportion of the skin diseases affecting neonates and infants. Clinically they are characterized by erythema, edema, scale, and sometimes crusts. The most common disorder is atopic dermatitis (AD) and, in fact, the term ‘eczema’ (which means ‘boiling over’) is often used by laymen to refer to AD. Seborrheic dermatitis and irritant dermatitis also frequently affect infants, while allergic contact dermatitis (ACD) is less common. Some nutritional, metabolic, and immunologic diseases have clinical manifestations that include eczematous eruptions which may be difficult to differentiate from AD, and they require a high index of suspicion for their occurrence. Atopic dermatitis is a chronic, pruritic skin condition with a frequently relapsing course. It was first described by Besnier in 1892 as ‘prurigo diasthétique’. In 1933, Wise and Sulzberger coined the term ‘atopic dermatitis’, and Hill and Sulzberger characterized the clinical entity 2 years later.1,2 AD is notable for inflammation, xerosis, crusts, excoriations, and lichenification, and it is the most common chronic inflammatory dermatosis of early childhood. The disease imposes an enormous burden on the personal, social, emotional, and financial resources of patients and their families. In the USA, annual direct costs for AD care are estimated at US$1–4 billion,3 which does not include the effects of lost productivity such as missed work and school days nor the impact on quality of life. The prevalence of AD has increased markedly over the past 50 years, with current rates of 15–29% in the USA, Europe, Japan, and other industrialized countries and lower but increasing rates in developing nations.4–6 AD affects individuals of all races, with several studies showing a slightly higher prevalence in black children.6 However, different countries with similar ethnic demographics have markedly different recorded prevalences, supporting the concept that environmental factors are important in disease expression.7 AD may be slightly more common in girls (female : male ratio of 1.3 : 1), although some studies note that it affects the sexes approximately equally.8 Multiple epidemiologic studies have examined risk factors for disease development. Only two factors show consistent, strong association: a family history of atopic diseases (i.e., asthma, allergic rhinitis, atopic dermatitis) and loss of function mutations in the filaggrin (FLG) gene. A positive family history of atopy is noted in approximately 70% of affected individuals. The odds of developing AD are 2–3-fold higher in children with one atopic parent, and this increases to as high as 3–5-fold if both parents are atopic.9,10 Some have noted a maternal history of AD itself to be more predictive.11,12 The FLG gene encodes a key protein involved in terminal differentiation of the epidermis, and its breakdown products are important in the formation of the skin barrier, including the stratum corneum, and natural moisturizing factor (see below). Loss of function mutations confer a risk for earlier-onset AD, as well as for more severe disease.13 AD is more frequent in higher social and socioeconomic classes.14 Other factors that may be associated with a greater risk for disease include small family size, migration from rural to urban environments, and increased parental education.7 Some have noted a higher incidence in association with maternal smoking and elevated birthweight, though not all studies corroborate these findings.15,16 The effect of exposure to pets in the home is unclear, due to conflicting data.17,18 The onset of disease is most commonly between 3 and 6 months of age, with approximately 60% of patients developing the eruption within the first year of life and 90% by 5 years of age.19,20 Atopic dermatitis is usually the first manifestation of the ‘atopic march,’ which refers to the subsequent development of food allergies, asthma, and allergic rhinitis, although this progression does not happen in all individuals and/or the sequence may vary.20 More recently, eosinophilic esophagitis and gastroenteritis have been added to the list of associated disorders.21 The predominant symptom is pruritus, which is difficult to control and a major cause of morbidity and poor quality of life for both the child and caregiver.22 It frequently interferes with sleep and can lead to complications such as secondary infection of excoriated lesions. Parents should be questioned about the infant scratching and rubbing against objects such as bedding or carpeting. Unfortunately, these actions further incite inflammation and the itch sensation, leading to a vicious cycle.23 There are three age-related phases to AD: infantile, childhood, and adult disease. During each phase both the sites of involvement and the morphology of the lesions change, although the phases often overlap. The infantile phase generally lasts until 2–3 years of age, the childhood phase from 2 years until puberty, and the adult phase from puberty onward. In infancy, AD characteristically begins on the cheeks (Figs 15.1, 15.2) and scalp (Fig. 15.3) and evolves over time to involve the lateral and extensor aspects of the arms and legs (Fig. 15.4). The periauricular areas are often affected and can lead to infra-auricular fissuring.24 While the trunk may have lesions, the diaper area is usually spared due to the retention of moisture and protection from rubbing and external triggers provided by these coverings (Figs 15.5, 15.6). Lesions are symmetric, scaly, erythematous patches and plaques, within which crusting is common. Acute lesions are more exudative. Lymphadenopathy may be noted and reflects local inflammation. During late infancy and the childhood phase, the flexural surfaces of the extremities become the most commonly involved sites, particularly the antecubital and popliteal fossae (Fig. 15.7). Other frequently involved locations include the neck, wrist and ankle flexures, and the creases between the thighs and buttocks. Lesions continue to be erythematous patches and plaques with crusting, but often there is increased excoriation. Chronic findings, such as lichenification (enhanced skin markings) and pigmentary changes start to become prominent (Fig. 15.8). The pigmentary alterations (hypo- or hyperpigmentation) often evoke parental concerns about scarring, although lesions generally do not leave scars unless severely infected or deeply excoriated. From puberty onwards, AD lesions tend to prefer the face, back, wrists, hands, and dorsal feet. There are a number of additional physical findings that, if present, aid in the diagnosis of atopic dermatitis (Box 15.1). These include other conditions associated with xerotic skin, such as ichthyosis vulgaris, keratosis pilaris, and pityriasis alba. Follicular lichenification with papules may be noted, especially in those with darker skin type. These are not specific to AD, however, and may be seen in otherwise healthy individuals as well. Other findings suggestive of an atopic diathesis that become more prominent with rubbing or scratching include an additional line or groove in the lower eyelid (the Dennie-Morgan fold), infraorbital darkening (‘allergic shiners’), and a prominent horizontal nasal crease (‘allergic salute’). The diagnosis of AD is made clinically. This is often more challenging in early infancy, when it can be difficult to distinguish AD from other infantile eczematous disorders. Family history is often helpful and absence of an atopic disorder in the family history makes the diagnosis much less likely, though does not preclude its occurrence. The most recognized diagnostic criteria are those proposed by Hanifin and Rajka in 1980, but the four major and 23 minor criteria are too numerous to be used routinely in clinical practice.25 Several expert groups have proposed more condensed sets of criteria,26–28 including the following,27 which is applicable to both children and adults: A. Essential features (must be present) a. Pruritus b. Eczematous dermatitis (acute, subacute or chronic) i. Typical morphology and age-specific patterns ii. Chronic or relapsing history B. Important features (seen in most cases, adding support to the diagnosis) b. Atopy i. Personal and/or family history ii. IgE reactivity c. Xerosis C. Associated features (these clinical associations help to suggest the diagnosis of AD but are too nonspecific to be used to define or detect AD for research or epidemiologic studies) a. Atypical vascular responses (e.g., facial pallor, white dermatographism, delayed blanch response) b. Keratosis pilaris/hyperlinear palms/ichthyosis d. Other regional findings (e.g., perioral changes/periauricular lesions) e. Perifollicular accentuation/lichenification/prurigo lesions. To date, there is no specific laboratory test to confirm the diagnosis of AD. Serum immunoglobulin E (IgE) is elevated in approximately 70–80% of patients, although even severe cases may have normal levels.23 The majority of patients also have increased circulating eosinophils. Recent discoveries of novel T-lymphocyte subsets and cytokine proteins have generated additional candidate markers. Thymus and activation-regulated chemokine (TARC, also called CCL17) is one such molecule suggested to increase with disease activity.29 However, to date, none have demonstrated adequate sensitivity and specificity to serve as a biomarker for the disease or its response to therapy. Laboratory testing is thus seldom needed in the routine evaluation of uncomplicated AD. Skin biopsies may be helpful in distinguishing AD from other inflammatory skin disorders, but the biopsy findings are not pathognomonic. The histology varies from an acute dermatitis with spongiosis and a perivascular lymphocytic infiltrate, to chronic changes in lichenified skin showing acanthosis, hyperkeratosis, and accumulation of Langerhans’ cells, macrophages, and mast cells in addition to lymphocytes around blood vessels. The superficial venular plexus has endothelial cell hypertrophy, basement membrane thickening, and increased numbers of immunoreactive nerve fibers.30,31 Eosinophils are not commonly present in acute AD, but are increased in number in chronic lesions. Mast cells may be normal to increased, and in different stages of degranulation. The pathogenesis of AD involves a complex interplay of genetic, immunologic, infectious, and other environmental factors. While still not completely understood, it appears to entail two major defective processes: dysfunction of the epidermal barrier and dysregulation of the immune system. Which process occurs first to initiate disease (‘outside-in’ vs ‘inside-out’) is an ongoing debate, although it is clear that they interact and exacerbate one another. This is compounded by a heightened response to environmental allergens, irritants, and microbial antigens.31,32 For many years, it has been known that AD involves genetic susceptibility, given familial clustering, concordance in monozygotic twins but discordance in dizygotic twins, and reports of transfer of AD by bone marrow transplantation.33,34 It has been most highly linked to genes of the epidermal differentiation complex located on chromosome 1q21, particularly the FLG gene, which is also the main cause of ichthyosis vulgaris. The association of FLG mutations with AD is one of the most robust known for complex genetic disorders.13 Large, population-based studies have identified over 35 mutations in affected individuals, with distinct mutations in different ethnic groups.35 As mentioned above, loss of function mutations are associated with early-onset and more severe, persistent disease, and additionally appear to give increased allergic sensitization and total IgE levels, a greater risk for asthma and food and other allergies, and a higher incidence of skin infections with herpes simplex virus (eczema herpeticum). But despite the strong association, FLG null mutations are seen in only approximately 18–48% of individuals with AD (depending on the population) and conversely, about 40% of individuals with such mutations do not develop the disease.13 Genetic screening of affected families has highlighted other chromosome regions that contribute to disease susceptibility, including cytokine and immune-related genes (e.g., those encoding interleukins (IL)-4, -5, and -13 and the regulated on activation of normal T-cell expressed and secreted (RANTES) chemotactic cytokine), genes encoding innate immunity receptors (e.g., the genes for toll-like receptors (TLR)-2 and -9), and genes implicated in other inflammatory diseases (e.g., those encoding nucleotide-binding oligomerization domain protein (NOD)-1 and NOD2).36,37 There are likely additional genes that remain to be discovered, and of those already identified to have high disease association, a number still require determination of exact function. Defects in both the structure and function of the epidermal barrier are present in AD. FLG mutations lead to disorganized keratin filaments and abnormal architecture of the lamellar bilayer and assembly of the cornified envelope. Decreased filaggrin breakdown products affect levels of natural moisturizing factor and the skin pH, resulting in increased transepidermal water loss and skin permeability. In addition, barrier function is markedly reduced due to a decline in ceramide levels, which are major water-retaining molecules, and from an imbalance of proteolytic enzymes that affect epidermal adhesion and their anti-protease counterparts.35,38 Abnormalities in tight junction expression that form a second barrier in the stratum granulosum further add to the pathology.34 These factors all lead to dry, xerotic skin, while facilitating entry of antigens through the damaged epidermis that stimulate cutaneous inflammation. Immunologic abnormalities are another major pathogenic mechanism. Antigens bind to and activate epidermal Langerhans’ cells and dermal dendritic cells. Circulating T cells that have increased expression of the cutaneous lymphocyte-associated antigen (CLA) receptor home to the skin, where they are primed by these antigen-presenting cells. In the acute phase of disease, these activated T cells overexpress cytokines of the Th2 class, notably IL-4 and IL-13, which mediate switching of immunoglobulin isotypes to IgE synthesis and upregulated expression of adhesion molecules on endothelial cells. IL-4 also stimulates mast cells to produce mediators such as histamine. IL-5 predominates in subsequent stages and is involved in eosinophil development and survival.31,39 While AD begins acutely as a Th2-mediated disorder, in its chronic phase it is characterized by Th0 cells (cells that share some activities of both Th1 and Th2 cells) and Th1 cells; associated cytokines include interferon-γ. The switch to Th0/1 cells involves the infiltration of the epidermis by inflammatory dendritic epidermal cells (IDEC) and production of IL-12 and IL-18, as well as several remodeling-associated cytokines. Also of significance is an increase in Th22 cells in chronic AD skin, which produce IL-22 that inhibits terminal differentiation and induces epidermal hyperplasia.40 Additional T cell subsets have been identified, but their roles in AD remain to be fully defined. T regulatory cells with normal immunosuppressive activity appear to be expanded in the peripheral blood of patients, but not in lesional skin or atopy patch test sites. Th17 cells have been found to be elevated in patients with acute AD, but not as prominently as in psoriasis.31 Enhanced allergen penetration through the damaged epidermis gives increased production of thymic stromal lymphopoietin (TSLP) by keratinocytes, which may be a critical link between the barrier and immune defects. TSLP has been shown to drive both the initial Th2 cytokine response and the switch to the Th0/1 phenotype.39 Mechanical injury such as scratching or rubbing, microbes, and proinflammatory cytokines themselves further stimulate release of TSLP, thus perpetuating the inflammation. The immunologic response contributes to additional barrier abnormalities, since IL-4 and IL-13 are known to suppress filaggrin expression and downregulate proper processing of profilaggrin. Antigens that gain entry include microbes. The antimicrobial barrier is compromised in AD from a combination of the defective physical barrier, an increased pH which may allow greater adherence and multiplication of bacteria, and toll-like receptor defects which diminish recognition of microbial agents. In addition, the increase in Th2 cytokines suppresses the synthesis of antimicrobial peptides by keratinocytes, specifically LL-37 (cathelicidin) and β-defensins 2 and 3.41 This predisposes atopic individuals to widespread skin infections due to bacteria and viruses (including herpes, molluscum, and vaccinia), and possibly to dermatophytes. Staphylococcus aureus, in particular, can trigger multiple inflammatory cascades. Staphylococcal toxins activate T cells in a superantigen-driven fashion and induce IgE-specific responses. These further skew the immune response toward Th2 cytokine production and explain the association of S. aureus infection with exacerbations of AD.42 The superantigens also cause a profound reduction in steroid responsiveness of T cells, giving another possible mechanism for disease flares. Exogenous protease inhibitors produced by S. aureus may also damage the epidermal barrier, thereby potentiating the absorption of allergens into the skin and leading to increased bacterial colonization. The impact of food exposure on the development of atopic dermatitis is controversial. Although it is known that food allergy and sensitization more commonly develop in children with atopic dermatitis, it is unclear how important food exposures are to the onset and course of atopic dermatitis, nor how common food-induced eczematous dermatitis is during infancy.43 Aeroallergens may also trigger immune responses, but sensitization is usually after infancy. The house-dust mite, Dermatophagoides pteronyssinus, has been cited in the pathogenesis of AD, but measures to decrease dust mite exposure are not necessarily protective.44 Malassezia yeasts have also been suggested but not confirmed to cause exacerbations of disease. The mechanisms underlying pruritus in AD are the subject of much investigation. In addition to histamine, several other mediators, such as neuropeptides, kinins, and cytokines, can induce pruritus. Studies have shown that the number of sensory and neuropeptide-containing nerve fibers is increased in lesional AD skin.30,45 Mast cells store large amounts of proteases, including tryptase, which may induce pruritoceptive itch when released in the proximity of these nerve fibers. These release stress-induced neuropeptides in parallel, which might activate mast cells through neurokinin receptors to perpetuate pruritic signals. The Th2 response contributes via increased levels of IL-31, which has recently been found to enhance pruritus. Staphylococcal superantigens also markedly enhance IL-31 production.45,46 The common disorders in the differential diagnosis of AD include seborrheic dermatitis, contact dermatitis, psoriasis, scabies, ichthyosis, tinea corporis, and keratosis pilaris (Box 15.2). In early infancy, seborrheic dermatitis may be virtually indistinguishable from AD due to the similarity in sites of involvement and morphology (Fig. 15.9). Irritant dermatitis is not uncommon, but allergic contact dermatitis is rare in the newborn period, although it may affect infants. A configuration suggesting an external source of exposure may be evident and helpful in determining the presence of a contact dermatitis. Psoriasis is not as common in infancy, but psoriasis vulgaris and pustular psoriasis may occur (see Chapter 16). In the case of psoriasis vulgaris, the diaper area is most commonly affected and consists of erythematous, well-demarcated plaques surmounted by some to little scale, which is less thick and silvery relative to other sites due to moisture and occlusion. About 5% of children appear to an overlap of psoriasis and atopic dermatitis lesions, and often have a family history of both disorders.47 Scabies may sometimes be difficult to distinguish from AD because both can cause severe pruritus, but it is rarely considered in the newborn, and is more relevant to infants. Here the face is not usually involved, whereas eczematous patches on the cheeks and xerosis are typical of AD. Moreover, the eruption of scabies has polymorphous lesions, including burrows, papules, nodules, eczematous and urticarial lesions, as well as pustules on the palms and soles. A recent onset of itching in family members is also helpful in differentiating the two conditions. When the skin lesions are associated with failure to thrive, diarrhea, infection, and/or signs in other organ systems, it is important to consider systemic disorders, such as nutritional and metabolic abnormalities, and genetic conditions including immunodeficiency (Box 15.3). Also in the differential diagnosis are proliferative disorders, most notably Langerhans’ cell histiocytosis, which often includes hemorrhagic lesions. Occasionally, AD may be so severe that the whole body becomes erythrodermic, causing confusion with other causes of erythroderma, particularly Netherton syndrome and nutritional deficiencies (see Chapter 18). Data from follow-up surveys show enormous variation in the long-term course and prognosis of AD, owing to differences in patient sampling techniques. Vickers48 reported that AD cleared by age 20 in 90% of patients, but the inclusion of infants with seborrheic dermatitis, which resolves within weeks to months, biased these results. Wüthrich49 found that of 121 infants followed to a mean of 23.5 years, 11% had resolution in childhood, an additional 25% cleared in adolescence, but 32% had a chronic continuous course from infancy into adulthood and 20% had reappearance of disease in adolescence. Despite these differences, there is a steady decline in AD prevalence over the childhood years. Up to 60–70% resolution of disease by adulthood appears to be a reasonable estimate, with an overall prevalence rate of 2–10% in adults.49,50 Early onset within the first 6 months of life, greater disease severity, and persistence into adolescence are predictors that the disease is more likely to continue into adult life.51 Other reported risk factors for adult disease include the presence of other atopic conditions such as asthma and allergic rhinitis, a family history of AD in parents or siblings, high serum IgE levels, and FLG null mutations in early-onset cases.49,51 It is, however, difficult to precisely predict if and when a particular child will have resolution or remission and management should continue until there is notable quiescence of lesions. But even after what appears to be resolution, easily irritated or dry skin may persist or hand eczema may affect the individual later in life.52 Figures on the incidence of asthma and allergic rhinitis following or in association with AD vary from 10–70%.51,53 In one survey, 36% suffered from allergic rhinitis, 28% from asthma, and 15% from both.54 There is an increased risk of developing asthma or AD if there is a family history of either condition. Higher rates have also been reported with more severe AD, with 60% having asthma and 62% with allergic rhinitis reported in one study.55 Mechanisms to link their occurrence are under study. Filaggrin mutations appear to increase the risk of development of asthma and other atopic conditions.13,53 It has been suggested that early or severe AD and epicutaneous sensitization to environmental allergens may lead to inflammation and development of allergic disease on re-exposure at other epithelial barrier sites (e.g., the respiratory or gastrointestinal tract). Systemic circulation of keratinocyte-derived TSLP may play an important role in this process.56 Although supported by animal models, this remains to be confirmed, along with whether prevention or early treatment of AD mitigates subsequent development of other atopic conditions. The management of AD should be directed at reducing signs and symptoms of disease and decreasing the frequency and severity of flares. It requires a multimodal approach that includes meticulous general skin care, hydration of the skin, avoidance of potential irritants and allergens, and reduction of pruritus and inflammation. Secondary infections should be treated and prevented. It is extremely important to educate the parents on the chronic nature of the disease, as many seek an immediate cure which is not possible. The goals of therapy, including the benefits, risks, and side effects of treatments, should be detailed. Because parents are inundated with much information, careful attention to detail, repetition of advice, and written handouts and action plans can improve treatment outcomes. Educational programs in the form of structured, multidisciplinary classes and nursing-led care are themselves effective primary interventions that can give some decrease in disease severity.57 Xerosis is a significant component of the disease, and hydrating the skin is a foundation of atopic dermatitis therapy. This is accomplished through frequent application of emollients or moisturizers and care in bathing techniques. The appropriate frequency of bathing is controversial, but limiting to a short duration using warm rather than hot water is recommended. Bathing alone leads to a decrease in hydration status of the skin once the water evaporates.58 On the other hand, there is evidence that bathing followed by the immediate application of an emollient while the skin is wet (the ‘soak and seal’ method) has an excellent hydrating effect, while removing scale and crust, and can be a tremendously useful treatment measure for patients during flares.59,60 The application of emollients and moisturizers independent of bathing can also improve hydration status, enhance barrier function, and be steroid sparing.59,61 They should be applied all over the body and after topical corticosteroids have been applied to lesional skin. There is no one preferred emollient or moisturizing agent. Ideally, it should be safe, effective, inexpensive, and free of additives, fragrances, and other potential sensitizing agents. Ointments are the thickest agents but creams and oils may also be used, while lotions should generally be avoided as they are less effective in decreasing xerosis. Products containing urea, lactic acid, and α-hydroxy acids may sting and their absorption in infants is unknown. Topical therapies have been developed to target skin barrier dysfunction and replace abnormal epidermal lipids. These include preparations having distinct compositions of lipids and ceramides to mimic endogenous lipids, and creams containing palmitoylethanolamide, hydrolipids, and/or filaggrin breakdown products.62 While they have shown some benefit and may be helpful as adjuvant agents, the few studies performed to date have not demonstrated superiority relative to traditional, less expensive options such as petrolatum ointment.62,63 But addressing the defective barrier is an important therapeutic concept, given the current understanding of AD pathogenesis. Topical corticosteroids were first introduced by Sulzberger and Witten in 1952 and remain a mainstay of AD treatment because of their excellent anti-inflammatory effects. They are grouped according to potency into seven classes, from very weak (VII) to superpotent (I). There is often fear and reluctance on the part of parents to use them, despite the extremely small number of reported side-effects in those treated, the majority of which have occurred in patients receiving inappropriate treatment.64,65 Physicians should encourage the correct use of topical corticosteroid preparations on affected inflamed areas. Both amount and potency are important and close monitoring during treatment is appropriate for safety and compliance. Precise information should be given to the parents regarding the manner of application, and written instructions are very useful. There are many methods of prescribing topical corticosteroids and other anti-inflammatory therapies. Published algorithms are available and may be used.66 Mild cases and/or facial or diaper areas are generally treated with low-potency steroids. For more significant flares, or if low-potency steroids are inadequate to control the disease, more potent topical corticosteroids are used for truncal and body surfaces. For severe flares, wet-wrap therapy (a damp layer of gauze or clothing with an outer dry layer) may be utilized with low- to mid-strength topical steroids for several days to increase their effect.67 Prescriptions should be given for sufficient treatment of the area involved, but with more caution and set limits with higher potency agents. In general, one finger-tip unit (the amount from the distal interphalangeal joint to the finger tip, ~0.5 g) should be applied over an area of two adult palms.64,68 For acute flares, areas should be treated until the inflammation disappears, with frequent and liberal use of emollients. Most should be used twice a day, although some agents are designated for once-daily use. Emollients should be continued after corticosteroids are discontinued and may help lessen disease recurrence and the need for anti-inflammatory agents. Systemic corticosteroids, particularly long-term use, are not indicated for the treatment of AD in neonates or infants. In addition to hypothalamic-pituitary-adrenal axis suppression, they can affect linear growth and have known rebound and other negative effects. Topical calcineurin inhibitors are another class of anti-inflammatory therapy for atopic dermatitis. In the USA, topical tacrolimus and pimecrolimus are approved as second-line agents for short-term and non-continuous chronic use in children aged 2 years and older, but they are sometimes used off-label. Unlike topical steroids, topical calcineurin inhibitors do not carry a risk of skin atrophy, and therefore may be helpful for sites of thinner skin (i.e., eyelids, face, and diaper area where steroid atrophy are a bigger concern) or to decrease the use of topical steroids in those with moderate or severe disease. There is histologic evidence that while both classes improve epidermal differentiation and decrease transepidermal water loss, topical steroids do not restore the epidermal barrier structures (e.g., lamellar body extrusion and lipid bilayer formation in the lower stratum corneum) to the same extent as calcineurin inhibitors.69 The most common side-effects with topical calcineurin inhibitors are local reactions such as stinging and burning, although this may improve after several applications or when first preceded by a short period of topical corticosteroid use. Patients should be monitored for signs and symptoms of cutaneous viral infections, although ongoing surveillance has not shown a consistent increase in frequency.70,71 These agents do carry a boxed warning stating that their long-term safety has not been established because of rare post-marketing cases of malignancy, specifically, of skin cancer and lymphoma. Interim analyses of ongoing, 10-year surveillance studies to address these concerns have not found evidence of increased malignancy rates relative to that expected in the general pediatric population.72,73 Several studies, including a large case–control study of 293 253 patients, have found an increased risk of lymphoma that correlates with AD severity, but not with topical calcineurin inhibitor use.74,75
Eczematous Disorders
Introduction
Atopic dermatitis
Risk factors
Clinical findings
History
Physical examination
Diagnosis
Etiology and pathogenesis
Differential diagnosis
Prognosis
Treatment
Xerosis/dry skin
Anti-inflammatory agents
Topical corticosteroids.
Topical calcineurin inhibitors.
Maintenance therapy.
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree