This article reviews the presentation of children with craniofacial anomalies by the most common sites of airway obstruction. Major craniofacial anomalies may be categorized into those with midface hypoplasia, mandible hypoplasia, combined midface and mandible hypoplasia, and midline deformities. Algorithms of airway interventions are provided to guide the initial management of these complex patients.
Key points
- •
The area of craniofacial skeleton involved in different syndromes is predictive of the airway problems typically encountered in affected patients.
- •
Most episodes of upper airway obstruction in children with craniofacial anomalies present in the immediate neonatal period.
- •
A majority of infants with Pierre Robin sequence (PRS) are able to be managed nonsurgically.
- •
The need for early endotracheal intubation is associated with an increased rate of subsequent surgical airway intervention.
- •
Tracheostomy rates are highest among children with combined midface and mandible hypoplasia.
Introduction
The medical management of children with craniofacial anomalies is complex. Therefore, their care is best addressed by a multidisciplinary team consisting of a geneticist, pediatrician, otolaryngologist, craniofacial surgeon, neurosurgeon, dentist, orthodontist, oral maxillofacial surgeon, audiologist, speech-language pathologist, and social services provider. The otolaryngologist plays a critical role in the evaluation and management of the airway in these children.
This patient population is uniquely predisposed to upper airway obstruction. Children with craniofacial anomalies often present with varying degrees of respiratory insufficiency during the neonatal period, requiring acute intervention. The airway management of these patients should be tailored to the degree and anatomic site of obstruction. Possible interventions range from positioning maneuvers to surgical airway establishment. This article presents a classification system of craniofacial anomalies by the site of airway obstruction, along with algorithms to guide the initial management of these patients.
Introduction
The medical management of children with craniofacial anomalies is complex. Therefore, their care is best addressed by a multidisciplinary team consisting of a geneticist, pediatrician, otolaryngologist, craniofacial surgeon, neurosurgeon, dentist, orthodontist, oral maxillofacial surgeon, audiologist, speech-language pathologist, and social services provider. The otolaryngologist plays a critical role in the evaluation and management of the airway in these children.
This patient population is uniquely predisposed to upper airway obstruction. Children with craniofacial anomalies often present with varying degrees of respiratory insufficiency during the neonatal period, requiring acute intervention. The airway management of these patients should be tailored to the degree and anatomic site of obstruction. Possible interventions range from positioning maneuvers to surgical airway establishment. This article presents a classification system of craniofacial anomalies by the site of airway obstruction, along with algorithms to guide the initial management of these patients.
Diagnostic evaluation
Bedside Clinical Assessment
All patients with suspected craniofacial anomalies should undergo a complete head and neck physical examination, along with flexible fiberoptic laryngoscopy. The physical assessment should focus on the cranial vault shape and suture patency, maxillomandibular relation, palatal clefting, tongue position, presence of stertor, presence of stridor, and overall work of breathing.
An endoscopic examination is most useful to determine the anatomic location of the obstruction. Nasal endoscopy should always be performed bilaterally to ensure patency of both nasal passages. Difficulty passing the endoscope through the nasal vestibule is diagnostic of pyriform aperture stenosis, whereas the inability to pass it more posteriorly suggests choanal atresia. In children with micrognathia, endoscopy can be used to confirm the presence of tongue-base obstruction.
Laboratory Studies
In select cases, laboratory studies may be helpful to diagnose subclinical respiratory insufficiency. Blood gas analysis can demonstrate elevated carbon dioxide levels, which is indicative of inadequate gas exchange and may predict impending hypercarbic respiratory failure.
Polysomnography
Prior studies have demonstrated a strong correlation between obstructive sleep apnea (OSA) and craniofacial anomalies. The overall incidence of positive OSA screening in children with craniofacial anomalies is 28.2%, but can affect at least 50% of children with particular craniosynostosis syndromes and facial clefts.
In the authors’ experience, however, polysomnography usually does not provide additional information that would alter management in the acute setting. The necessity of early airway intervention is most often a clinical decision based on a patient’s degree of respiratory compromise and feeding ability, and is unrelated to sleep.
Craniofacial syndrome classification
Major craniofacial anomalies may be categorized into those with midface hypoplasia, mandible hypoplasia, combined midface and mandible hypoplasia, and midline deformities. This approach provides an anatomic classification of the most common craniofacial anomalies ( Table 1 ).
| Site | Syndrome |
|---|---|
| Midface | Apert Carpenter Crouzon Down Pfeiffer Saethre-Chotzen |
| Mandible | PRS Nager Stickler |
| Combined | Bilateral hemifacial microsomia Treacher Collins syndrome |
| Midline | Choanal atresia Midline cleft Pyriform aperture stenosis |
Midface hypoplasia
Pathophysiology
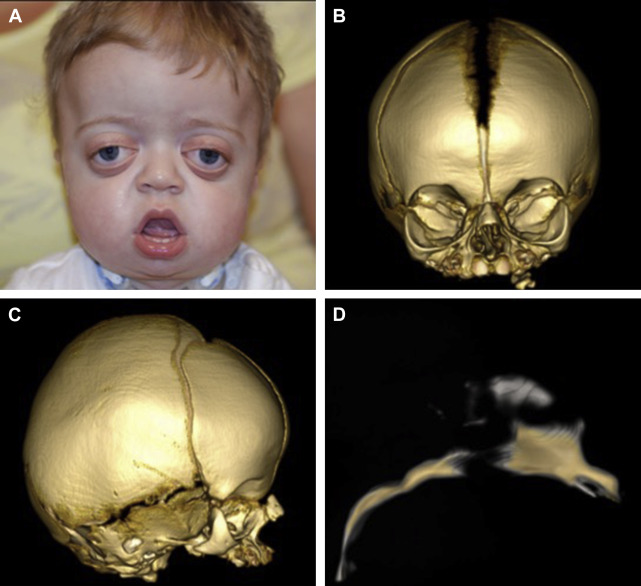
Many craniosynostosis syndromes involve premature fusion of the cranial sutures with concomitant midface hypoplasia. The midfacial growth tends to occur more slowly and arrests prior to 10 years of age, resulting in a short anterior cranial base, acute cranial base angle, and class III occlusion. Additionally, the maxilla is constricted and highly arched, which may impinge on the vertical dimension of the nasal cavity.
These anatomic abnormalities, along with the normal development of lymphatic tissue within the Waldeyer ring, leads to nasopharyngeal and oropharyngeal obstruction. The degree of obstruction can be so severe that it mimics bilateral choanal atresia during presentation. In addition to a routine physical examination, all children with suspected craniosynostosis syndromes should undergo a nasopharyngeal evaluation with passage of a 5/6 French suction catheter or fiberoptic endoscope.
CT is the initial imaging modality of choice to evaluate the bony anatomy of the midface. Three-dimensional reconstructions can also be obtained to plan for future craniofacial surgery ( Fig. 1 ). Finally, MRI is indicated to rule out associated central nervous system abnormalities, such as hydrocephalus or Chiari malformation.
Airway Management Techniques
A previous study demonstrated that approximately half of patients with midface hypoplasia require some form of airway intervention. Medical therapy with topical vasoconstrictors represents a minimally invasive intervention that can relieve mild nasal obstruction, particularly during periods of concurrent infection.
Nasopharyngeal airways, also known as nasal trumpets, offer another potential method to relieve midface obstruction. In the authors’ experience, however, placement of a nasopharyngeal airway is not always possible nor beneficial in these patients. Nasopharyngeal airways themselves are space occupying and, therefore, compromise the cross-sectional area of the nasal passage. They are also prone to crust formation and may incite further mucosal inflammation. If a nasopharyngeal airway is implemented, frequent saline irrigation with gentle suctioning should be performed to maintain its patency.
Noninvasive positive pressure ventilation may serve as a short-term method of ventilation in patients with respiratory compromise not requiring emergent intubation. The presence of midface hypoplasia and exorbitism can interfere, however, with the fit of a face mask. Should bag-valve mask ventilation be necessary, it is recommended that the mouth be held in an open position while firmly applying pressure on the face mask to form a seal.
Acute airway obstruction not responsive to noninvasive measures should be managed with orotracheal intubation. Orotracheal intubation usually does not pose a unique challenge in this patient population, unless there are other sites of airway obstruction or vertebral abnormalities limiting neck extension. Conversely, nasotracheal intubation may be difficult and require the use of a smaller endotracheal tube. The need for initial endotracheal intubation is predictive of an increased rate of subsequent airway intervention.
The final step of the management algorithm for upper airway obstruction is tracheostomy. The overall incidence of tracheostomy among children with craniofacial anomalies is approximately 20%. In a recent retrospective cohort study, craniofacial anomalies were cited as the second most common reason for infant tracheostomy.
Children with midface hypoplasia and resultant nasal and/or nasopharyngeal obstruction commonly require tracheostomy. Infants who undergo tracheostomy placement secondary to craniofacial anomalies generally have a long period of cannulation with a mean length of 6.7 years. There is evidence, however, that long-term pediatric tracheostomy is associated with morbidity and mortality. Potential complications of a tracheostomy include
- •
Pneumothorax or pneumomediastinum
- •
Accidental decannulation
- •
Cannula obstruction
- •
Hemorrhage
- •
Infection
- •
Stoma maintenance issues
- •
Tracheal stenosis
- •
Speech and swallowing impairment
Finally, caregivers of children with tracheostomy tubes experience a significant burden with respect to the child’s illness severity and associated costs.
Mandible hypoplasia
Pathophysiology
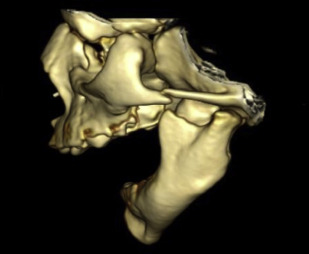
Mandible development begins during the fourth week of gestation with neural crest cell migration to the head and neck region. Mandible hypoplasia is thought to be the result of inadequate neural crest cell incorporation into the first branchial arch. Congenital hypoplasia usually occurs bilaterally. More than 60 syndromes with mandible hypoplasia have been identified to date.
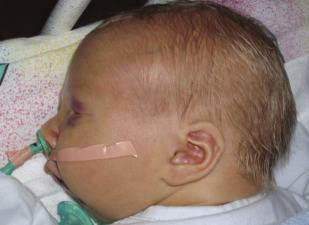
Pierre Robin sequence refers to the triad of micrognathia, glossoptosis, and a U-shaped cleft palate ( Fig. 2 ). It is considered a sequence because multiple anomalies are caused by a series of in utero events initiated by a single anomaly. The initial event, mandible hypoplasia, positions the tongue posterosuperiorly in the oral cavity between the palatal shelves. This abnormal tongue position then causes a mechanical disruption in palatal closure with resultant cleft formation. Approximately 20% to 40% of PRS cases are isolated and the remainder occur as part of a syndrome, such as Stickler syndrome.
Airway Management Techniques
Airway interventions for children with mandibular hypoplasia aim to reposition the tongue anteriorly and alleviate pharyngeal obstruction. Approximately three-fourths of these patients require an airway intervention. Options for the management of upper airway obstruction in this patient population are both nonsurgical and surgical ( Table 2 ).
| Intervention | |
|---|---|
| Nonsurgical | Positioning Nasopharyngeal airway Endotracheal intubation |
| Surgical | Glossopexy procedures Mandibular distraction osteogenesis Tracheostomy |
The most conservative treatment of mandibular hypoplasia involves prone or decubitus positioning. Previous studies have shown high success rates with positioning maneuvers alone for the management of PRS. The long-term efficacy of such positioning techniques, however, has not been demonstrated.
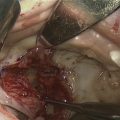
If a child with mandibular hypoplasia continues to experience mild obstructive episodes despite prone positioning, nasopharyngeal airway placement should be performed. A nasopharyngeal airway maintains a lumen between the tongue base and posterior pharyngeal wall during periods of collapse. Published success rates of nasopharyngeal airway placement range from 48% to 100%. Most children require continuation of the nasopharyngeal airway for several weeks to months, but its use has been described up to 27 months without complication. Often, a nasopharyngeal airway is used in the short term until a more definitive intervention is completed ( Fig. 3 ).