Syndromic craniosynostosis affects up to 1:30,000 live births with characteristic craniofacial growth restrictions, deformities, and other associated abnormalities, such as carpal-pedal anomalies and cognitive function impairment. More than 150 syndromes are associated with craniosynostosis. This article describes some commonalities and distinguishing features and management of syndromic synostosis. Also addressed is secondary synostosis, which is often found in syndromic children with problems related to microcephaly, hydrocephalus, or shunt-induced craniosynostosis, although pathophysiologically and genetically different. The importance of obtaining a thorough history and a complete physical and examination is highlighted. Adjuvant testing and multidisciplinary management are discussed.
Key points
- •
Syndromic craniosynostosis is rare, occurring in 1:30,000 to 1:100,000 live births.
- •
Fibroblast growth factor receptor and tumor growth factor-β receptor mutations have been reported to be associated with many forms of syndromic craniosynostosis.
- •
Intracranial hypertension, developmental delays, and strabismus are more frequent in syndromic forms of craniosynostosis than isolated synostosis.
- •
Distraction osteogenesis is a useful adjunct in syndromic synostosis to increase intracranial volume and is helpful with fronto-orbital and midface advancements.
- •
Addressing decompression by increasing intracranial volume and decreasing intracranial pressure before 1 year of age is a common goal through an interdisciplinary team approach.
Introduction
Syndromic craniosynostosis, premature fusion of the cranial sutures in syndromic patients, has been reported to affect from 1:100,000 to 1:30,000 live births causing restriction in skull and skull base growth with associated midface hypoplasia and dysmorphisms being common. There are greater than 150 syndromes associated with craniosynostosis. Patients with syndromic craniosynostosis are more liable to have ventricular expansion, hydrocephalus, expanded subarachnoid space, and cerebellar tonsillar herniation compared with patients with sporadic single-suture synostoses. Increased intracranial pressure (ICP) is more likely to occur in patients with syndromic craniosynostosis and multisuture synostosis. Cerebrospinal fluid (CSF) flow disturbance may present in one of two ways: ventriculomegaly present before craniofacial surgery, or normal or small ventricles that progressively increase after forehead advancement and cranial suture release. If these conditions are progressive, they can be associated with decreased cognitive function and intelligence quotients. Chronic transependymal fluid transfer, ventriculomegaly causing deformity of long tracks, and periventricular fibrosis are likely to change brain function. In syndromic synostosis, brain developmental anomalies, such as septum pellucidum and corpus callosum dysgenesis, and hypogyria are not infrequent. Brain distortion and local compression causes additional damage. Crouzon syndrome has the highest incidence of hydrocephalus. In Apert syndrome, ventricular dilation occurs frequently, but usually is nonprogressive in nature. In most cases of syndromic craniosynostosis, venous outflow obstruction and/or chronic tonsillar herniation are found. In cases of venous hypertension, a higher CSF pressure is required to maintain the CSF outflow balance. Cognitive impairment is common in Apert syndrome and cloverleaf deformities presenting 20% of the time in Crouzon, Pfeiffer, and Saethre-Chotzen type I syndromes.
Timing of surgery for patients with syndromic craniosynostosis depends on the presenting symptoms, such as the degree of exorbitism and signs and symptoms of elevated ICP. There is a higher incidence of hydrocephalus following early surgery, which is one consideration for delaying surgery. The incidence of hydrocephalus in craniosynostosis is 4% to 10%. There is no clear consensus on the ideal operative window for syndromic craniosynostosis; however, delaying surgery beyond 1 year results in a higher likelihood of elevated ICP, cognitive deficits, and behavioral problems. Therefore, decompression, increasing intracranial volume, and addressing ICP before 1 year of age is a common goal. One institutional study states that when there is no concern for elevated ICP the ideal operative window for these procedures in the syndromic population seems to be 6 to 9 months of age. Pediatric ophthalmology evaluation of vision, strabismus, and fundoscopic examination to rule out papilledema are important. When papilledema is present, MRI and/or ICP monitoring are often useful in evaluating signs of pressure or hydrocephalus. Proptosis is common as a result of hypoplastic orbits frequently leading to scleral show, lagophthalmos, and dry eyes and may require either temporary tarsorrhaphies or fronto-orbital advancement (FOA) to protect the eyes from exposure keratitis and eventual midface advancement to greater improve orbital volume.
Chiari malformation–related central apnea or alteration of cranial nerve nuclei–related muscle tone might contribute to obstructive sleep apnea (OSA). Obstructive airway can lead to brain hypoperfusion and hypoxemia. Chiari malformation can occur because of obstruction of venous outflow and reduced posterior fossa volume. Cinalli and coworkers in 1995 revealed that premature lambdoid suture closure is the main factor for Chiari malformation and proved that this pathologic process is present almost universally in Crouzon syndrome. Based on the authors experience, idiopathic or brain-related multisuture synostosis can present the same way once synostosis is present with increase in brain volume.
There is greater likelihood with syndromic patients to require multiple surgeries, not only to improve cranial volume and shape early on but also to address midface, orbital, and cranial deformities that often persist and/or recur as a result of poor bone growth in early childhood and adolescence. Diagnosing and treating upper airway obstruction and timely decompression and reconstruction of the skull are all crucial in syndromic patients.
Introduction
Syndromic craniosynostosis, premature fusion of the cranial sutures in syndromic patients, has been reported to affect from 1:100,000 to 1:30,000 live births causing restriction in skull and skull base growth with associated midface hypoplasia and dysmorphisms being common. There are greater than 150 syndromes associated with craniosynostosis. Patients with syndromic craniosynostosis are more liable to have ventricular expansion, hydrocephalus, expanded subarachnoid space, and cerebellar tonsillar herniation compared with patients with sporadic single-suture synostoses. Increased intracranial pressure (ICP) is more likely to occur in patients with syndromic craniosynostosis and multisuture synostosis. Cerebrospinal fluid (CSF) flow disturbance may present in one of two ways: ventriculomegaly present before craniofacial surgery, or normal or small ventricles that progressively increase after forehead advancement and cranial suture release. If these conditions are progressive, they can be associated with decreased cognitive function and intelligence quotients. Chronic transependymal fluid transfer, ventriculomegaly causing deformity of long tracks, and periventricular fibrosis are likely to change brain function. In syndromic synostosis, brain developmental anomalies, such as septum pellucidum and corpus callosum dysgenesis, and hypogyria are not infrequent. Brain distortion and local compression causes additional damage. Crouzon syndrome has the highest incidence of hydrocephalus. In Apert syndrome, ventricular dilation occurs frequently, but usually is nonprogressive in nature. In most cases of syndromic craniosynostosis, venous outflow obstruction and/or chronic tonsillar herniation are found. In cases of venous hypertension, a higher CSF pressure is required to maintain the CSF outflow balance. Cognitive impairment is common in Apert syndrome and cloverleaf deformities presenting 20% of the time in Crouzon, Pfeiffer, and Saethre-Chotzen type I syndromes.
Timing of surgery for patients with syndromic craniosynostosis depends on the presenting symptoms, such as the degree of exorbitism and signs and symptoms of elevated ICP. There is a higher incidence of hydrocephalus following early surgery, which is one consideration for delaying surgery. The incidence of hydrocephalus in craniosynostosis is 4% to 10%. There is no clear consensus on the ideal operative window for syndromic craniosynostosis; however, delaying surgery beyond 1 year results in a higher likelihood of elevated ICP, cognitive deficits, and behavioral problems. Therefore, decompression, increasing intracranial volume, and addressing ICP before 1 year of age is a common goal. One institutional study states that when there is no concern for elevated ICP the ideal operative window for these procedures in the syndromic population seems to be 6 to 9 months of age. Pediatric ophthalmology evaluation of vision, strabismus, and fundoscopic examination to rule out papilledema are important. When papilledema is present, MRI and/or ICP monitoring are often useful in evaluating signs of pressure or hydrocephalus. Proptosis is common as a result of hypoplastic orbits frequently leading to scleral show, lagophthalmos, and dry eyes and may require either temporary tarsorrhaphies or fronto-orbital advancement (FOA) to protect the eyes from exposure keratitis and eventual midface advancement to greater improve orbital volume.
Chiari malformation–related central apnea or alteration of cranial nerve nuclei–related muscle tone might contribute to obstructive sleep apnea (OSA). Obstructive airway can lead to brain hypoperfusion and hypoxemia. Chiari malformation can occur because of obstruction of venous outflow and reduced posterior fossa volume. Cinalli and coworkers in 1995 revealed that premature lambdoid suture closure is the main factor for Chiari malformation and proved that this pathologic process is present almost universally in Crouzon syndrome. Based on the authors experience, idiopathic or brain-related multisuture synostosis can present the same way once synostosis is present with increase in brain volume.
There is greater likelihood with syndromic patients to require multiple surgeries, not only to improve cranial volume and shape early on but also to address midface, orbital, and cranial deformities that often persist and/or recur as a result of poor bone growth in early childhood and adolescence. Diagnosing and treating upper airway obstruction and timely decompression and reconstruction of the skull are all crucial in syndromic patients.
Evaluation and diagnosis of craniosynostosis
Detailed pregnancy history, birth history, family history, and medication or drug exposure in utero is crucial to document. Syndromic forms of synostosis are often inherited in an autosomal-dominant (AD) fashion, although spontaneous mutations frequently occur. A review of systems should note any presence of associated headaches, irritability, seizures, or neurodevelopmental or cognitive delays. A timeline of the duration and progression of head shape change is important because progressive unilateral occipital flattening suggests deformational plagiocephaly, whereas bilateral progressive flattening implies positional brachycephaly. Bilateral coronal craniosynostosis is one of the most common findings in syndromic synostosis including Crouzon, Muenke, and Apert with a head that is short in the anteroposterior dimension (brachycephaly) frequently coupled with increased vertical height in the cephalocaudal dimension of the skull (turricephaly). A good history is often all that is needed to distinguish the typical positional brachycephalic skull, a shape that progressively developed, from syndromic types of brachycephaly.
However, deformities present since birth are more suggestive of synostosis, although positional deformities can also be present at birth as a result of malpositioned fetal lie, oligohydramnios, and in utero constraints of pregnancy with twins or multiples. Photographic documentation from a variety of views of the head and face including frontal, left lateral, right lateral, vertex, and oblique views should be obtained ( Fig. 1 ).
Meticulous physical examination of the head includes palpation of the sutures and fontanelles, inspection of the orbits and face (noting any asymmetry, ptosis, lagophthalmos, dystopia, hypertelorism, hypotelorism, facial paresis/paralysis), measurement of the fronto-occipital circumference, and calculation of the cephalic index ratio (skull width/skull length). Location, size, and shape of the ears relative to the head should be noted. A head to toe examination is essential for evaluation of syndromic craniosynostosis to detect carpal/pedal anomalies ( Table 1 ) that may be present and the presence of possible cardiac or respiratory difficulties; when there is still uncertainty a three-dimensional (3D) computed tomography (CT) of the head definitively rules in or out craniosynostosis.
| Condition | Craniofacial Phenotype | Gene a |
|---|---|---|
| Loeys-Dietz syndrome (aortic aneurysm, arterial tortuosity, hypertelorism, cleft palate/bifid uvula, craniosynostosis) | Craniosynostosis, cleft palate, hypertelorism | TGF β R1 TGF β R2 |
| Apert syndrome | Craniosynostosis (brachycephaly); wide midline defect closes by coalescence of bony islands; midface malformations; dental crowding; cleft or narrow palate with swellings | FGFR2 |
| Beare-Stevenson cutis gyrata syndrome | Craniosynostosis (kleeblattschädel) or cloverleaf skull | FGFR2 |
| Boston-type craniosynostosis | Craniosynostosis (kleeblattschädel), forehead retrusion, frontal bossing | MSX2 |
| Cleidocranial dysplasia | Delayed suture closure, frontal parietal bossing, wormian bones, hyperdontia, tooth eruption defects | RUNX2 |
| Craniofrontonasal syndrome | Craniosynostosis (brachycephaly), central defect between frontal bones, hypertelorism, divergent orbits | EFNB1 |
| Crouzon syndrome | Craniosynostosis (brachycephaly), pronounced digital impressions of skull, midface hypoplasia, shallow orbits | FGFR2 |
| Crouzon syndrome with acanthosis nigricans | Craniosynostosis | FGFR3 |
| Greig cephalopolysyndactyly | Craniosynostosis in small percentage of cases, frontal bossing, sagittal ridging, hypertelorism | GLI3 |
| Muenke-type craniosynostosis (nonsyndromic) | Craniosynostosis (brachycephaly) | FGFR3 |
| Parietal foramina | Symmetric parietal bone defects, cleft lip/palate | MSX2, ALX4 |
| Parietal foramina with cleidocranial dysplasia | Symmetric parietal bone defects | MSX2 |
| Pfeiffer syndrome | Craniosynostosis (brachycephaly) | FGFR1, FGFR2 |
| Saethre-Chotzen syndrome | Craniosynostosis (especially brachycephaly), flat forehead, low hairline | FGFR2, TWIST1 |
| Shprintzen-Goldberg (marfanoid) syndrome | Craniosynostosis (especially lambdoid and sagittal sutures), maxillary and mandibular hypoplasia, palatal abnormalities | SKI |
| Thanatophoric dysplasia II | Craniosynostosis (cloverleaf skull/kleeblatschädel deformity) | FGFR3 |
a Gene abbreviations that have not been defined/spelled out are not acronyms or abbreviations.
Midface or mandibular hypoplasia, and adenotonsillar hypertrophy if present, can contribute to sleep-disordered breathing or OSA. CT and MRI of the head are often useful to identify any hydrocephalus, signs of ICP elevation, or posterior fossa problems, such as Chiari ( Fig. 2 ), which could lead to central sleep apnea.
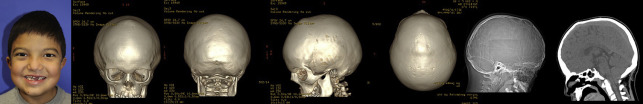
Elevation of ICP can present acutely, subacutely, or chronically from microcephaly, venous anomalies, and hydrocephalus. Nonspecific signs and symptoms such as irritability, lack of appetite, and inconsolable crying are important indicators of increased ICP. More prominent signs are a bulging fontanelle and engorged scalp veins.
The authors agree with a growing body of craniofacial and neurosurgeons that posterior cranial distraction (PCD) is a minimally invasive way to slowly improve intracranial volume with potential to decrease mild to moderate elevations of ICP and help normalize the brachycephalic head shape by growing the skull in the anteroposterior axis. It is thus often ideal in syndromic craniosynostosis involving bicoronal or multisuture synostosis with brachycephaly and/or microcephaly.
Craniofacial team
Syndromic craniosynostosis often involves multiple organ systems so an interdisciplinary team approach is now the standard of care for these complicated patients. This team can include a plastic surgeon, pediatric otolaryngologist or facial plastic surgeon trained in craniofacial surgery, a pediatric neurosurgeon, an oral-maxillofacial surgeon, a pediatric anesthesiologist and intensivist, a pedodontist, orthodontist, a prosthodontist, a pediatric ophthalmologist, a psychologist, a geneticist, an audiologist, a speech pathologist, and pediatrician.
Distraction osteogenesis
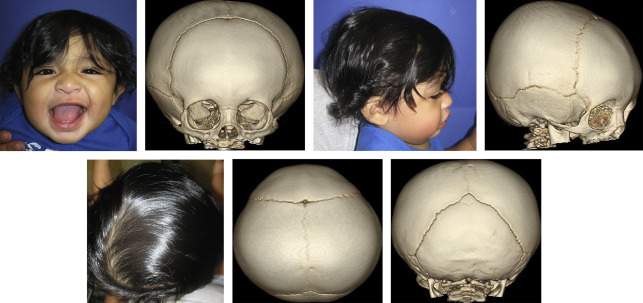
Distraction osteogenesis (DO) can be used to advance the mandible, midface, orbits, and skull including lower level and higher level Le Fort osteotomies, PCD, or fronto-facial (Monobloc) advancements using either external or internal distractors ( Fig. 3 ). Cranial distraction has been used to increase microcephalic skulls or provide added volume in the case of shunt-induced craniosynostosis. It has also been described for use across patent sutures and other more unconventional total cranial vault reconstruction in a recent paper describing 285 different cases of coronal, metopic, lambdoid, and sagittal synostosis, of which 33 were syndromic and 28 were secondary synostosis. Typically an open bicoronal approach is used because eventual anterior cranial vault reconstruction (ACVR) with FOA and frontal cranioplasties are usually required months after distraction, although in cases of secondary synostosis, microcephaly, or persistent ICP elevation despite shunt, the approach can be accomplished through several small incisions and endoscopic assistance.
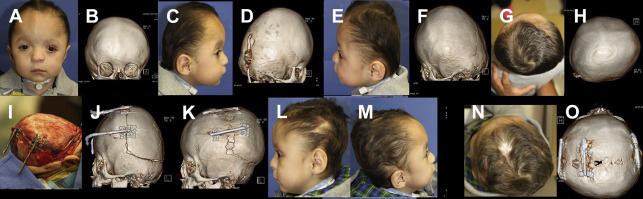
Following this approach, craniotomies are planned, marked, and performed either with a craniotome, Kerrison rongeurs, osteotomes, or peizosurgery device. Two to four single-vector cranial distractors are placed in the desired plane, typically anteroposterior for posterior vault distraction. The incisions are closed and after a latency period (few days to a week), depending on the age of the patient, activation of the distraction commences at a typical rate of 0.5 mm twice daily. A commonly used internal device for PCD, made by KLS Martin (Jacksonville, FL), is 30 mm in length and has a switch that, once toggled, prevents parents from accidentally incorrectly turning the distractors in the wrong direction. Additionally, a ratcheting screw provides an audible and tactile click with each turn of the activating wrench/driver, giving parents additional feedback that they are correctly using the wrench and that the distraction is preceding appropriately. The desired volume is achieved after 3 to 5 weeks and a portion of the external distractor arms is thereafter removed in the office. After another 2 to 3 months of allowing the bony consolidate to calcify and mature, the patient is returned to the operating room for removal of distractors and at times concomitant ACVR. This approach is particularly useful in syndromic patients with brachycephaly in whom adequate advancement is difficult to achieve through conventional surgery.
Transsutural DO (TSDO) has been described in which a simple suturectomy of the pathologic suture or even placement of distractors across patent sutures is used to distract the skull. TSDO potentially allows for greater cranial vault expansion compared with traditional posterior cranial vault reconstruction or ACVR, although it adds increased complexity and potential hardware complications by virtue of using greater number of distractors.
One TSDO study showed shorter mean operative time compared with most prior studies. This results in decreased bleeding and transfusion volumes when compared with total cranial vault reconstruction with bone grafting. TSDO has also been used with some success for total calvarial remodeling by removal of all sutures. However, in this study approximately 50% developed Chiari malformations immediately after surgery and 50% with ocular abnormalities. The authors of this article have no experience with TSDO.
Theoretic advantages of distraction are that the soft tissue can be slowly stretched allowing for greater amounts of increased intracranial volume gained with lesser risk of relapse or tight scalp closures that are subject to dehiscence, scar widening, and exposure of hardware or bone grafts. In addition to improving the aesthetic appearance, there is functional benefit by giving the brain space ( Fig. 4 ).