Mode of inheritance
Gene
Target protein
AD = autosomal dominant
AR = autosomal recessive
Dominant DEB
Dominant DEB, generalized
AD
COL7A1
Type VII collagen
Dominant DEB, acral
AD, AR
COL7A1
Type VII collagen
Dominant DEB, pretibial
AD, AR
COL7A1
Type VII collagen
Dominant DEB, pruriginosa
AD, AR
COL7A1
Type VII collagen
Dominant DEB, nails only
AD
COL7A1
Type VII collagen
Dominant DEB, bullous dermolysis of the newborn
AD, AR
COL7A1
Type VII collagen
Recessive DEB
Recessive DEB, generalized severe (former recessive DEB-Hallopeau-Siemens)
AR
COL7A1
Type VII collagen
Recessive DEB, generalized intermediate
AR
COL7A1
Type VII collagen
Recessive DEB, inversa
AR
COL7A1
Type VII collagen
Recessive DEB, localized
AD, AR
COL7A1
Type VII collagen
Recessive DEB, pretibial
AD, AR
COL7A1
Type VII collagen
Recessive DEB, pruriginosa
AR
COL7A1
Type VII collagen
Recessive DEB, centripetalis
AR
COL7A1
Type VII collagen
Recessive DEB, bullous dermolysis of the newborn
AR
COL7A1
Type VII collagen
The most severe DEB form is termed recessive DEB, generalized severe (RDEB-gen sev), which is molecularly characterized by complete loss of collagen type VII expression. Generalized blistering of an extremely fragile skin starts at birth, both spontaneously and secondary to often minor mechanical forces. Large lesions typically arise on trauma-exposed sites or over bony prominences and heal with milia formation (i.e., keratin-filled cysts, arising when keratin has become trapped under the epidermis), hypo- or hyperpigmentation, and atrophic scarring (Fig. 42.1). Further cutaneous features include occasionally multiple angiomas.
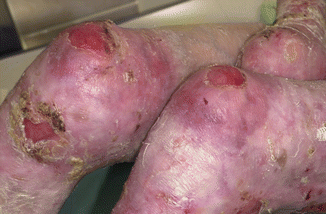
Fig. 42.1
Partly crusted erosions and atrophic scarring in the knee region of a patient with generalized severe recessive dystrophic EB
Repeated blistering and excessive scarring on hands and feet may result in total encasement of digits and acral mitten deformities (pseudosyndactyly) which is a clinical hallmark of recessive dystrophic EB (RDEB-gen sev; RDEB-inversa [RDEB-I]; infrequently in dominant DEB and rarely observed in generalized severe junctional EB) (Fig. 42.2) [2, 3]. Pseudosyndactyly initially presents as partial fusion (proximal webbing-adhesion, synechiae formation) within the interdigital spaces and is followed by progressive bridging and complete fusion of all of the individual digits to a keratinaceous cocoon-like, scarred mass (mitten deformities). Contractures of fingers, toes, hands (adduction contractures of thumb), and feet begin to develop as early as within the first year of life in RDEB-gen sev and RDEB-I (infrequently in DDEB). Also proximal contractures may occur, especially within the popliteal and antecubital fossae and axillary vaults. Muscle atrophy, bone absorption, and progressive functional disablement (difficulties in weight bearing, standing, walking; reduced fine manipulative skill; loss of digital prehension) including wheelchair dependency are dramatic sequelae.

Fig. 42.2
Mitten deformities on feet (a) and pseudosyndactyly as well as contractures and complete anonychia of both hands (b) in generalized severe recessive dystrophic EB
Involvement of the nail apparatus causes peri- or subungual blistering, hemorrhages, and nail bed hyperkeratosis with onycholysis and onychomadesis. Early nail dystrophy and loss are triggered by trauma (thus, great toenails are more often severely affected) and correlate with disease severity and progression [4]. Symptoms are often just a mild cosmetic problem, but sometimes also the cause of severe disability. Periodic nail shedding and regrowth results in progressive onychodystrophy (shortened, thickened, dome-shaped, yellow-brown nail plate) and onychogryphosis (thickened, opaque, yellow, oyster-like nail plate). Mitten deformities, nail atrophy (very thin, brittle, short), and, ultimately, complete loss due to nail bed and matrix damage by repetitive blistering and scarring occur.
Increased hair fragility and sparse hair as well as scarring alopecia secondary to trauma, blistering, and/or infections involving interfollicular epidermis and upper portions of the hair follicle are constant features of RDEB-gen sev that are further aggravated by traction [5]. Anemia and sepsis may account for a telogen effluvium.
EB nevi are a frequent phenomenon in RDEB-gen sev as in other types of EB. They develop as most common acquired melanocytic nevi, beginning as flat, black to brown pigmented lesions, which later, while acquiring dermal components, lose their pigment (Fig. 42.3). Gradually appearing in infancy or adolescence as stippled maculae, these moles develop papillomatous areas over years, resulting in dermal shagreen-like nevi [6]. EB nevi typically arise in sites of previous bullae or erosions, often with a darker rim at the confines of the preceding vesiculation. This led to the hypothesis that pathogenetically the repetitive disruption of the basement membrane primes local nevus cell nests or single melanocytes to break senescence and undergo proliferation [7, 8]. Viable melanocytes/nevus cells, probably deriving from incipient nevi or subclinical nests of nevus cells, free-float in the fluid-filled cavity of EB blisters (“flocking-bird melanocytes”) and, after settling down at random (often trapped in the sharp angel at the edge of the blister), proliferate excessively in the microenvironment of epidermal regeneration (involving cytokines and growth factors such as hepatocyte growth factor, interleukin-8, granulocyte-macrophage colony-stimulating factor, prostaglandin E2 or leukotriene B4) [9]. The arbitrary arrangement of independently proliferating melanocytic clones and enhancing secondary changes due to wound healing, scar formation, disruption of rete ridges, and neovascularization probably account for the irregular appearance of these moles.
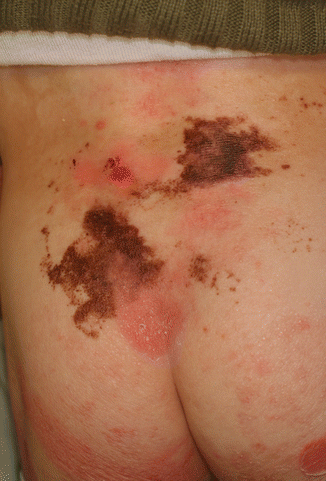
Fig. 42.3
EB nevus. Large, polycyclic pigmentary lesion with satellites in a patient with generalized severe recessive dystrophic EB
Thereby EB nevi frequently manifest with clinical, histological, and dermoscopic features highly suggestive of melanoma [10]. Although the state of chronic skin wounding and regeneration seems to promote cancerogenesis (as demonstrated for squamous cell carcinomas and melanomas [11–14]), their course is usually benign and spontaneous disappearance can occur [10, 15]. The observation of malignant transformation of an EB nevus to invasive melanoma in one case of EB simplex [16] nevertheless underscores the necessity to maintain a high index of suspicion for melanoma and a low threshold to biopsy of suspicious (morphologically changing) moles.
Skin-derived squamous cell carcinoma (SCC) is a very common complication of particularly RDEB-gen (> RDEB, generalized intermediate > RDEB-I and JEB) (Fig. 42.4). It is occasionally also observed on the tongue or in the esophagus [2]. Tumors arise most commonly at sites of chronic wounding, regeneration, or scarring and as early as within the second decade of life. Their frequency further increases thereafter. This is complicated by a very aggressive course and extremely high rates of metastasis as well as recurrence. Referring to solid statistics provided by the US National EB Registry, metastatic SCC is the primary cause of death in RDEB, occurring in the majority of patients with RDEB-gen sev. The cumulative risk of developing SCC and subsequent death in patients with RDEB-gen sev at age 55 is greater than 90 and 78 %, respectively [17, 18].
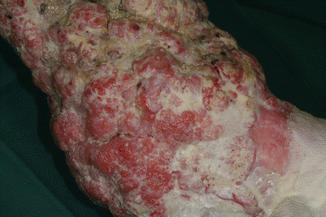
Fig. 42.4
Squamous cell carcinoma on the left lower leg of a patient with generalized intermediate recessive dystrophic EB that presents as large ulcerated keratinized tumor masses adjacent to a chronic ulcer. These tumors are highly aggressive and have extremely high rates of metastasis
Risk and short latency appear to parallel severity, extent, and persistence of ulceration, wound healing response, and scarring, in turn correlating with intrinsic loss of type VII collagen expression and decreased or absent anchoring fibrils [19, 20].
Although data suggest repetitive tissue stress and remodeling, growth activation of keratinocytes, polymorphisms of matrix metalloproteinases, and reduced activity of natural killer cells to promote malignant transformation either as a predisposing microenvironment or a distinct pathology, the exact pathogenic pathways involved in tumorigenicity hitherto remain largely unknown.
RDEB-gen sev patients additionally face a possibly increased age-matched risk for melanoma in childhood (2.5 % by age 12 compared to 1.35–2.7 % lifetime risk in general population) [15, 17]. Other (internal) malignancies do not develop more frequently in any subtype of inherited EB as compared to the general population [2]. Epidemiologic data, however, is limited because of largely missing comprehensive cohort studies.
In recessive DEB, generalized intermediate, blistering is less severe (although continuing to develop throughout life), and mutilating deformities do not develop, as the mostly underlying compound heterozygous mutations account for (functionally and/or structurally) residual protein expression. The clinical picture, however, is variable, with some patients displaying widespread disease, while in others blisters are limited primarily to the extremities (Fig. 42.5). Skin lesions heal invariably with scars and milia. The risk for the development of squamous cell carcinomas is increased.
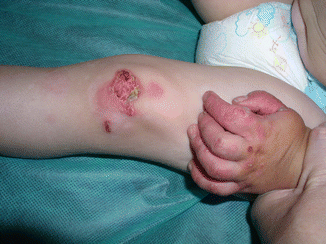
Fig. 42.5
Generalized intermediate recessive dystrophic EB. Blisters and crusted erosions particularly at trauma-exposed sites like the knee and hand
Very rare localized subtypes of recessive DEB include the following:
In RDEB-I, blistering is predominantly located in intertriginous but also lumbosacral, acral, and axial areas. Further observed are nail dystrophy; involvement of oral cavity and gastrointestinal and genitourinary tract; stenosis of meatus acusticus externus; anemia; and growth retardation.
Pretibial RDEB is characterized by pretibial blistering at birth or early infancy, involvement of hands and feet, lichen planus-like skin lesions, nail dystrophy [may precede skin blistering], excessive caries, and constipation. It may be associated with localized cutaneous amyloid deposition derived from degenerated keratinocytes secondary to scratching or induced by damage to epidermal-dermal junction/blister formation. Presenting with fragile blisters and erosions that are often overshadowed by pruritic lichenified plaques, this variant is often misdiagnosed as lichen amyloidosis [21].
Localized RDEB presents with blistering on hands and feet from early infancy on. Nail dystrophy is described.
RDEB pruriginosa: generalized or localized blisters, erosions, milia, and atrophic scars usually from infancy on, folliculitis-like lesions on scalp [22], and nail dystrophy. Excessive pruritus on a background of inherited skin fragility leads to skin signs resembling acquired inflammatory disorders such as hypertrophic lichen planus or prurigo nodularis; onset may not occur until adult life, further compounding difficulties in distinguishing between inherited or acquired skin disorders [23].
RDEB centripetalis: pretibial and acral blistering at birth or from early infancy on, nail dystrophy, and involvement of oral mucosa.
RDEB: bullous dermolysis of the newborn which presents with generalized blistering from birth or early infancy on, milia, atrophic scarring, and dystrophic nails.
Dominant DEB patients have an autosomal dominantly inherited altered type VII collagen expression. The coexistence of a defective as well as wild-type allele, leaving some anchoring fibrils functionally intact, accounts for a generally milder phenotype. Blistering starts at birth or soon thereafter, ranging from mainly acral involvement to disseminated lesions and scarring. “Albopapuloid” lesions may be present, i.e., small grouped hypopigmented papules usually on the lower back (Fig. 42.6). It was speculated whether they result from a reactive accumulation of (immature) collagen and amorphous glycosaminoglycan in EB skin or represent connective tissue nevi, respectively [24, 25]. Disease activity commonly diminishes with advancing age.
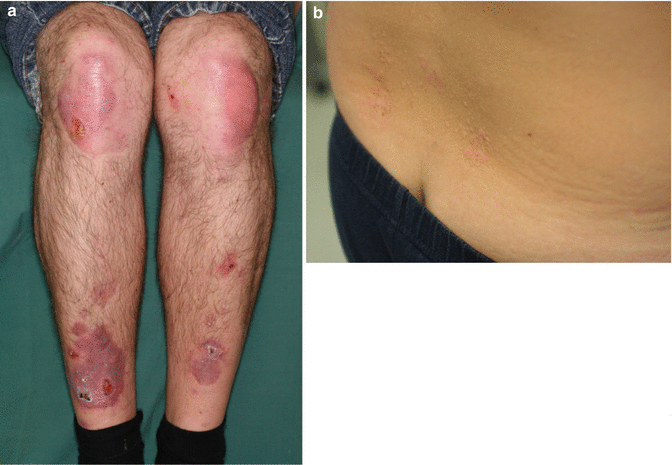
Fig. 42.6
Dominant dystrophic EB presenting with moderate scarring, erosions, and crusts at mechanically exposed acral sites (a) and with “albopapuloid” lesions, i.e., small grouped hypopigmented papules on the lower back (b)
Nail abnormalities with dystrophy or loss at birth or infancy may be an isolated finding as reported in DDEB [26, 27]. This “nails-only DDEB” variant is a newly recognized subtype of DEB, in which nail dystrophy is the only clinical feature (Fig. 42.7). The deformity is often limited to the toenails and can be mild and thus easily overlooked. Moreover, nail involvement without blistering may be present for generations before a DEB family member develops blisters in the skin. Diagnosis should thus be considered in patients with an autosomal dominant trait of toenail dystrophy, even when there is no history of blistering [28].

Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 Dermatitis Herpetiformis
Dermatitis Herpetiformis

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


