Disorders with Photosensitivity
Moise Levy M.D.
Kurt Hirschhorn M.D.
Judith Willner M.D.
Leonard Milstone M.D.
Clinical Pearls
(ML)
(KH)
(JW)
(LM)
Bloom Syndrome
Inheritance
Autosomal recessive; RecQL3 helicase gene on 15q26.1
Prenatal Diagnosis
Amniocentesis: amniotic fluid cell culture reveals high number of sister chromatid exchanges
DNA analysis
Incidence
Over 100 case reports; increased frequency amongst Ashkenazi Jews from eastern Europe; M:F=1.3:1
Age at Presentation
First few months of life
Pathogenesis
A mutation in the RecQL3 gene, encoding a DNA helicase responsible for unwinding DNA, interferes with DNA replication and repair leading to increased sister chromatid exchanges and chromosomal breaks, gaps, and rearrangement; shares helicase family mutation with Werner’s syndrome, xeroderma pigmentosum (XP) B, XPD, and Rothmund-Thomson syndrome
Key Features
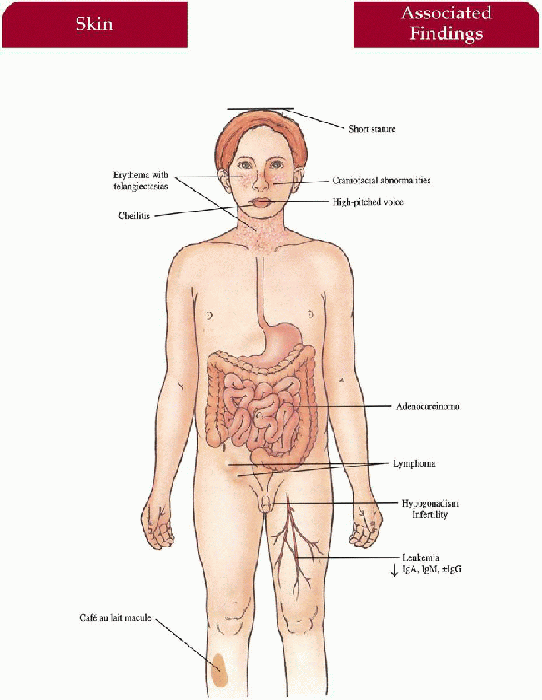
Skin
Photodistributed erythema with telangiectasias in butterfly distribution on nose and cheeks; eruption may involve the ears, forearms and dorsal hands; with/without
bullae
Cheilitis
Café au lait macules
Craniofacial/Body Habitus
Long, narrow face with prominent nose, malar hypoplasia, small mandible; short stature
Ear-Nose-Throat
High-pitched voice
Immunology
Decreased immunoglobulin (Ig) A, IgM, with/without IgG with recurrent respiratory and gastrointestinal infections
Endocrine
Hypogonadism, infertility (males)
Neoplasia (20%)
Acute leukemia, lymphoma, and GI adenocarcinoma most common
Differential Diagnosis
Cockayne syndrome (p. 242)
Rothmund-Thomson syndrome (p. 238)
Lupus erythematosus
Erythropoietic protoporphyria (p. 224)
Laboratory Data
DNA analysis
Chromosome analysis
Immunoglobulin levels
Management
Referral to dermatologist—diagnosis, sun protection
Referral to pediatric infectious disease specialist, hematologist/oncologist, endocrinologist—antibiotics, carcinoma surveillance, short stature management respectively
Prognosis
Increased risk of premature death (second to third decade) due to malignancy; otherwise good general health with infections, skin changes decreasing with age
Clinical Pearls
In infancy, they have severe failure to thrive … In terms of nailing the diagnosis, the immunoglobulin abnormalities and a test for chromosome instability are most useful … There are labs set up to run the chromosome tests at the National Institutes of Health (NIH) and Armed Forces Institute of Pathology … They can be bothered by the facial erythema … I generally give families the names of cosmetic coverups and send them to our equivalent of Bloomingdale’s … Photoprotect with sunscreens, hats, frogskin clothing. ML
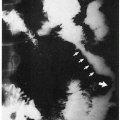
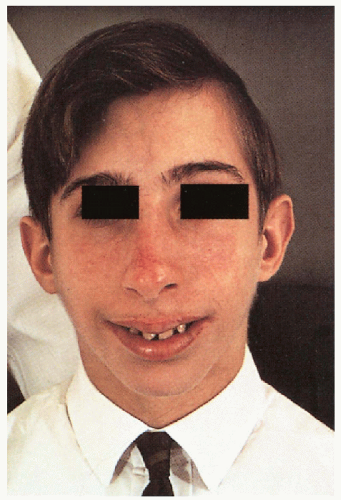 8.1. Boy with erythema, telangiectasias in butterfly distribution on nose and cheeks with characteristic facies. (66) |
 8.2. Affected brother and sister with similar cutaneous changes and facies. (66) |
|
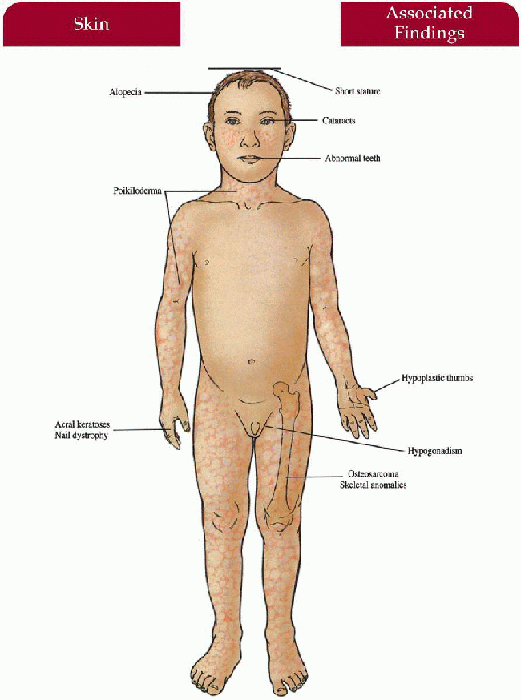
Rothmund-Thomson Syndrome
Synonym
Poikiloderma congenitale
Inheritance
Autosomal recessive; RecQL4 helicase gene on 8q24 in some cases
Prenatal Diagnosis
DNA analysis
Incidence
Over 130 cases reported; F>M; increased with consanguinity
Age at Presentation
Three to 6 months old (cutaneous changes)
Pathogenesis
A mutation in RecQL4 helicase gene contributes to phenotype in some cases with predicted DNA repair problems and susceptibility to cancers as seen in Werner, Bloom and XPB, XPD (other helicase family gene mutation syndromes); otherwise unknown defect
Key Features
Skin
Initial erythema, edema on face rapidly replaced by red-brown reticulated patches associated with atrophy, hypopigmentation, telangiectasias on face, buttocks, extensor extremities
Photosensitivity with/without bullae
Acral verrucous keratoses after puberty—may precede squamous cell carcinoma
Hair
Alopecia of scalp, eyebrows, eyelashes
Nails
Dystrophic nails (25%)
Musculoskeletal
Short stature, small hands and feet, hypoplastic/absent thumbs, variety of skeletal abnormalities
Eyes
Juvenile cataracts (40% to 50%)—begins at 3 to 7 years old
Endocrine
Hypogonadism (25%)
Teeth
Dental dysplasia
Neoplasia (rare)
Reports of osteosarcoma, fibrosarcoma, and squamous cell carcinoma
Differential Diagnosis
Bloom syndrome (p. 234)
Cockayne syndrome (p. 242)
Werner syndrome (p. 158)
Kindler syndrome
Laboratory Data
Long-bone x-rays
Management
Referral to dermatologist—diagnosis, photoprotection
Referral to ophthalmologist—yearly screen and cataract management
Referral to orthopedist, dentist, endocrinologist, hematologist/oncologist if symptomatic
Prognosis
If no malignancy then normal life span; usually normal intelligence
Clinical Pearls
Poikilodermatous changes are not necessarily confined to the sun-exposed areas … One patient I followed had striking involvement of the buttocks, another over the vulva … We had one child with such severe bowing of her distal tibias that the radiologist initially called and asked if the patient was wheelchair-bound … She had required multiple osteotomies to keep her mobile … Both of the patients referred to above have died from osteosarcoma. It is imperative to follow the bone changes for such degeneration. Malignant risks appear particularly marked in patients with documented RECQL4 mutations … Have also seen many adults with hyperkeratosis involving palms/soles, which can be marked and clinically significant … Half the children have subtle learning disabilities … All the patients I follow have similar facies … Baseline ophthalmologist examination and then yearly… I counsel about photoprotection. ML
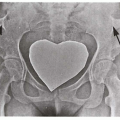
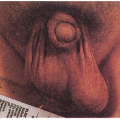
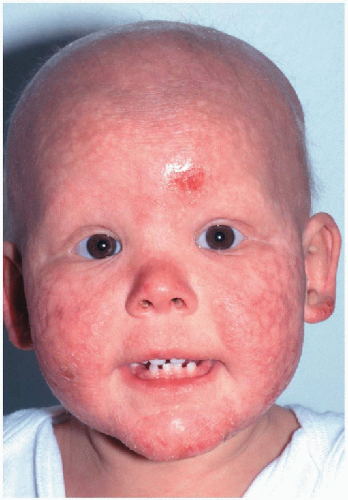 8.3. Child with poikilodermatous changes of the face, forehead erosion, and dental dysplasia. (91) |
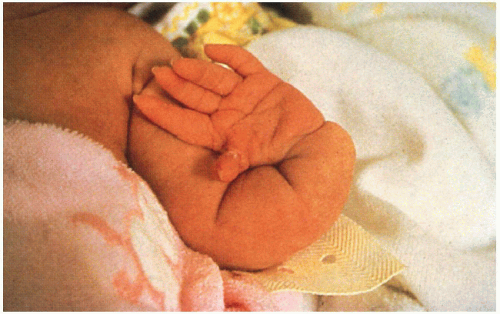 8.4. Congenitally absent radius with hypoplastic thumb. (92) |
|
Cockayne Syndrome
Inheritance
Autosomal recessive; Cockayne syndrome group A (CSA): ERCC8 gene on chromosome 5
Cockayne syndrome group B (CSB): ERCC6 gene on 10q11
Prenatal Diagnosis
Amniocentesis/amniotic fluid cell culture—deficient RNA synthesis and increased cell death after UV irradiation
DNA analysis
Incidence
Very rare; M=F; CSB most common (80% of cases)
Age at Presentation
Birth to 2 years old; some later, into teens
Pathogenesis
Mutations in ERCC8 and ERCC6 impairs DNA repair in active genes specifically, rendering the patient hypersensitive to UV and leads to progressive neurodegeneration; overlap of XPB, XPD, XPG with Cockayne exists in small number of patients
Key Features
Skin
Photosensitive eruption with erythema and scale in “butterfly” distribution on face—may resolve with hyperpigmentation and atrophy
Subcutaneous fat loss on face with resultant sunken eyes, aged appearance
Craniofacial/Body Habitus
Cachectic dwarf with microcephaly, thin nose, large ears (“Mickey Mouse” appearance); disproportionately long limbs with joint contractures; large, cold hands and feet
Nervous System
Diffuse demyelination of the central nervous sytem (CNS) and peripheral nerves with progressive neurologic deterioration; mental retardation; intracranial calcifications
Ear-Nose-Throat
Sensorineural deafness
Eyes
“Salt-and-pepper” retinal pigment, miotic pupils may be difficult to dilate, cataracts, optic atrophy
Teeth
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


