Disorders with Immunodeficiency
Moise Levy M.D.
Clinical Pearls
(ML)
Wiskott-Aldrich Syndrome
Inheritance
X-linked recessive; Wiskott-Aldrich Syndrome (WAS) gene on Xp11
Prenatal Diagnosis
DNA analysis
Fetal blood sample in male fetus—abnormally small platelets
Incidence
1:250,000; males only
Age at Presentation
First few months of life with bleeding problems
Pathogenesis
Mutation in WAS gene that encodes WASp, a protein important in lymphocyte and megakaryocyte signal transduction and actin filament assembly, impairs T-cell activation and natural killer cell function
Key Features
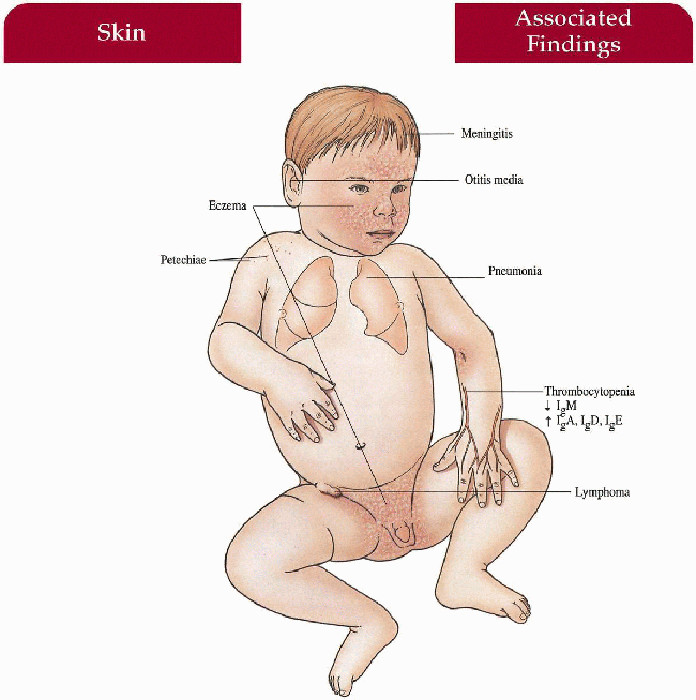
Skin
Atopic dermatitis with increased involvement on scalp, face, flexures; secondary bacterial infection, eczema herpeticum, molluscum contagiosum, lichenification
Blood
Thrombocytopenia with petechiae, purpura, epistaxis, bloody diarrhea, hematemesis, intracranial hemorrhage
Infectious Disease
Recurrent bacterial infections (especially encapsulated organisms) with otitis media, pneumonia, meningitis, septicemia
Increased susceptibility to HSV (eczema herpeticum), Pneumocystis carinii, human papilloma virus
Immunology
Increased immunoglobulin (Ig) A, IgD, IgE, and decreased IgM
Impaired cell-mediated and humoral immune response
Increased IgE-mediated urticaria, food allergies, asthma
Neoplasm
Lymphoreticular malignancy (20%)—non-Hodgkin’s lymphoma most common
Differential Diagnosis
Atopic dermatitis
Severe combined immunodeficiciency (SCID) (p. 264)
Hyper-IgE syndrome (p. 262)
Chronic granulomatous disease (p. 258)
Laboratory Data
Complete blood count (CBC) with differential, platelets, mean platelet volume (MPV)
Serum Ig levels
Immunoblot/fluorescence-activated cell sorter analysis (FACS): WASp protein expression in mononuclear cells from peripheral blood
DNA analysis
Management
Bone marrow transplant (BMT)
Splenectomy with long-term antibiotic prophylaxis
Appropriate antibiotics, intravenous immunoglobulin, plasma/platelet transfusions
Topical corticosteroids, moisturizers, prophylactic oral acyclovir
Referral to hematologist/oncologist, infectious disease specialist, and dermatologist
Prognosis
Frequently, premature death in first decade of life because of infection > hemorrhage > malignancy
Clinical Pearls
The bloody nature of the diarrhea may help distinguish from SCID… The big problem with platelet transfusions is that the patient may develop platelet antibodies, making subsequent transfusions less useful… Standard atopic care with vigorous use of moisturizers first and foremost… Defer topical steroids unless necessary… Crust with eczema can be hemorrhagic… Failure to thrive can be seen as with all primary immunodeficiencies in infancy. Often, early death without BMT although longer survival without BMT may occur… ML
|
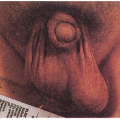
 9.1. Eczematous dermatitis accentuated in flexures. Note splenectomy scar. (1) |
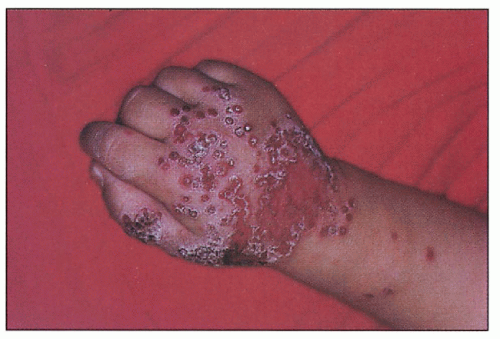 9.2. Eczema herpeticum on dorsum of patient’s hand. |
Chronic Granulomatous Disease
Inheritance
X-linked recessive (76%)—gp91-phox (phagocyte oxidase) gene on Xp21.1; autosomal recessive (24%)—p47 (more common), p67-phox genes on 7q1 1, 1q respectively
Prenatal Diagnosis
DNA analysis
Fetal blood sample—nitroblue tetrazolium (NBT) reduction assay of fetal leukocytes
Incidence
Approximately 1:250,000 to 500,000; M:F=9:1
Age at Presentation
Birth to 1 year old
Pathogenesis
Genetically heterogeneous group of immunodeficiency disorders caused by phox mutations in the nicotinamide dehydrogenase phosphate (NADPH) oxidase enzyme complex leading to an inability to produce a respiratory burst and defective killing of catalase positive organisms within phagocytic leukocytes; (respiratory burst occurs when NADPH oxidase acts as a catalyst for the production of superoxides and ultimately microbicidal oxidants)
Key Features
Skin
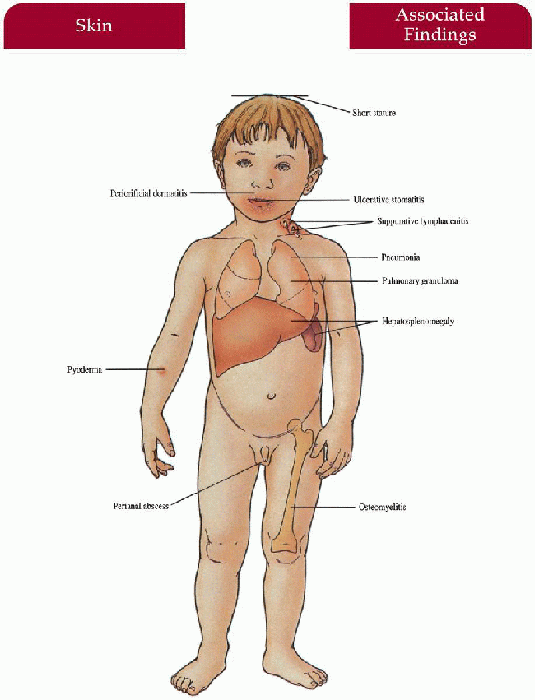
Recurrent pyoderma (Staphyloccus aureus most common), periorificial dermatitis with purulent drainage and regional lymphadenopathy, abcesses (perianal most common), granulomas
Mucous Membranes
Ulcerative stomatitis, chronic gingivitis
Lymph Nodes
Suppurative lymphadenitis with abscesses and fistulas (cervical nodes most common)
Lungs
Pneumonia with abscesses, cavitations, empyema (Staphylococcus, Aspergillosis, Nocardia)
Gastrointestinal Tract
Hepatosplenomegaly with granulomas, abscesses, chronic diarrhea, malabsorption
Musculoskeletal
Osteomyelitis (serratia marcescens most common), short stature
Differential Diagnosis
Hyper-IgE syndrome (p. 262)
SCID (p. 264)
Chédiak-Higashi syndrome (p. 62)
B-lymphocyte disorders
Laboratory Data
NBT reduction assay: leukocytes unable to reduce dye—no blue color change
CBC, erythrocyte sedimentation rate (ESR), immunoglobulin levels, chest x-ray, delayed hypersensitivity—skin test normal
Lungs, liver, bone imaging—locate occult inflammation; bacterial cultures
Immunoblot analysis of defective NADPH enzymes; DNA analysis
Management
Referral to infectious disease specialist—antibiotics
Referral to surgery—debridement, drainage, access to deeper infections; systemic steroids for obstructive visceral granulomas
Referral to dermatologist—topical and oral antibiotics, topical corticosteroids, antibacterial cleansers
Leukocyte transfusions, subcutaneous gamma interferon, BMT
Gene therapy for p47-phox form has been attempted with some persistence of corrected leukocytes at 6 months
Identify carriers and evaluate for lupus-like syndrome
Prognosis
Variable life span depending on control of infections; most with normal life span but poor quality of life
Clinical Pearls
A couple of kids have had resistant bacterial scalp infections… I have never seen the periorificial dermatitis… NBT is a simple test to do… Can be done right in the doctor’s office… May pick up tender joints by observing the kid’s positioning of the arms and legs… One-quarter of a cup of clorox to a full bath of water is a useful, cheap way to keep up with recurrent skin infections… I tend to not restrict patient’s activity… Subcutaneous gamma interferon three times a week has provided clinical improvement… Patients with chronic granulomatous disease (CGD) tend to be better targets for prophylactic antibiotics… ML
 9.3. Three-month-old boy with granulomatous scalp nodule which grew serratia marcescens. (96) |
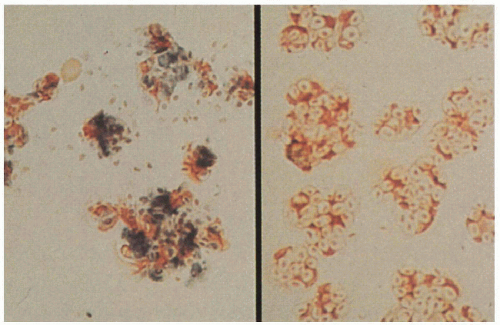 9.4. Left. Nitroblue tetrazolium (NBT) reduction assay with normal control demonstrating leukocytes’ ability to reduce dye and produce blue color change. Right. Abnormal leukocytes in affected patient unable to reduce dye. (97) |
|
Hyper-Immunoglobulin E Syndrome
Synonym
Job syndrome thought to be a variant
Inheritance
Autosomal dominant with variable expressivity; chromosome 4q21-gene unknown
Prenatal Diagnosis
None
Incidence
Rare—approximately 150 patients described; M=F
Age at Presentation
First few months to first year of life
Pathogenesis
Impaired regulation of IgE function and deficient neutrophil chemotaxis may play a role in susceptibility to infection
Key Features
Skin
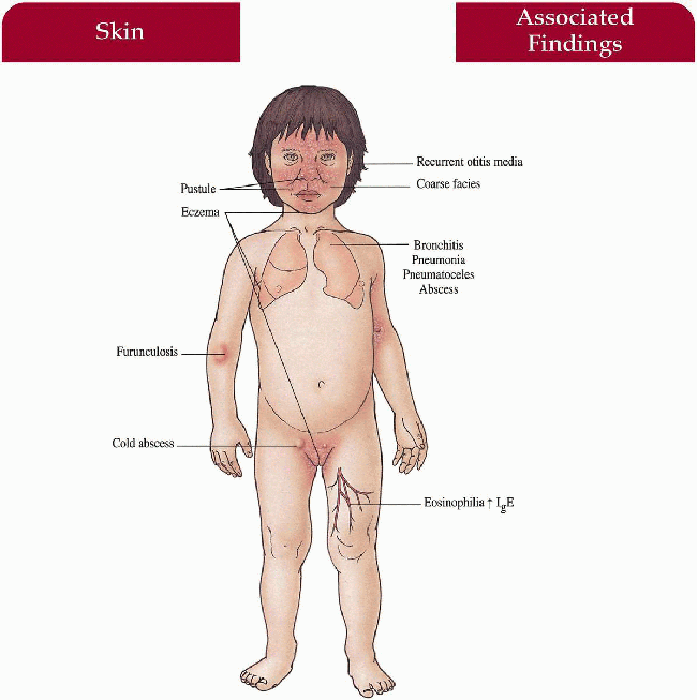
Excoriated papules, pustules, furuncles, cellulitis and abscesses (30% cold) on scalp, neck, axillae, groin, periorbital; paronychial infection; infected with S. aureus (most commonly); also Candida, Streptococcus
Eczematous dermatitis increased in flexures, postauricular, hairline
Sinopulmonary
Recurrent bronchitis, lung abscesses, pneumonia secondary to S. aureus, Haemophilus influenzae; pneumatoceles with bacterial/fungal superinfection, empyemas, recurrent otitis media, sinusitis
Craniofacial
Coarse facies with broad nasal bridge, prominent nose
Musculoskeletal
Osteopenia with secondary fractures (pelvis, long bones, ribs most common), scoliosis, hyperextensible joints
Dental
Retained primary teeth, lack of development of secondary teeth
Laboratory Data
IgE level markedly increased, IgD increased
Abnormal leukocyte/monocyte chemotaxis in some cases
Peripheral eosinophilia
Bacterial cultures
Management
Long-term antistaphylococcal antibiotics for prophylaxis, therapy; incision/drainage of abscesses; antifungal therapy
Interferon-γ and γ-globulin—improve chemotaxis and decrease IgE levels, respectively
Cimetidine—immune modulation
Referral to thoracic surgeon—excision of persistent pneumatoceles
Prognosis
Death may occur at early age if persistent bacterial or fungal infection of lungs exist; otherwise, with prophylaxis, prognosis is excellent
Clinical Pearls
I lump this syndrome with Job syndrome… You’re talking about a child with diffuse dermatitis and deep-seated pyogenic infection … Not just your casual impetigo but deep-seated abscesses, chronic severe ear infections… Remember atopics can have IgE in the thousands as well… This is typically not a syndrome of infancy… Large pneumatoceles will be amenable to lobectomy… ML
|
 9.5. Coarse facies with broad nasal bridge, extensive dermatitis. (98)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|