Disorders of Porphyrin Metabolism
Vincent DeLeo M.D.
Clinical Pearls
(VD)
Porphyria Cutanea Tarda (PCT)
Inheritance
Autosomal dominant; Uroporphyrinogen (UROGEN) decarboxylase gene on 1p34—less common (20% of cases)
Sporadic/acquired—more common
Prenatal Diagnosis
DNA analysis in familial cases
Incidence
Most common porphyria; approximately 1:25,000 in North America; M=F
Age at Presentation
Usually third to fourth decade of life; some familial patients in first decade of life
Pathogenesis
UROGEN decarboxylase gene mutation leads to UROGEN decarboxylase deficiency in erythrocytes, hepatocytes, and 50% of all tissues in familial form; deficient enzyme in hepatocytes in sporadic form
Increased uroporphyrin (URO) in skin leads to photosensitization after absorbing light energy in soret band (400-410 nm)
Alcohol, estrogen, hepatic tumors precipitate all acquired forms and may unmask familial cases
Key Features
Skin
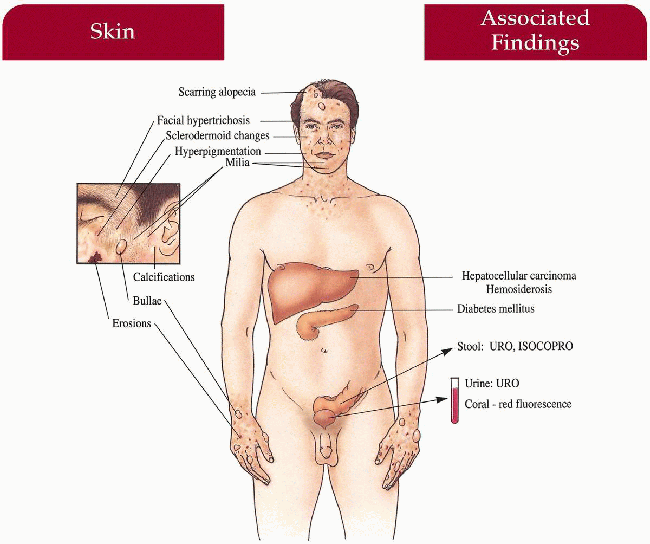
Delayed-type photosensitivity with bullae, erosions, skin fragility, facial hypertrichosis, hyperpigmentation; late changes include scarring, milia, sclerodermoid changes, subcutaneous calcification, alopecia
Gastrointestinal
Hepatocellular carcinoma (rare)
Liver hemosiderosis
Endocrine
Diabetes mellitus
Differential Diagnosis
Variegate porphyria (VP; p. 218)
Pseudoporphyria
Epidermolysis bullosa acquisita
Hereditary coproporphyria (p. 221)
Laboratory Data
Plasma porphyrin level and fluorescence spectrum—24-hour urine porphyrin level (≠URO) complete blood count (CBC), liver function tests, iron level, hepatitis panel, liver scan; in-office urine screen—coral pink fluorescence with Wood’s light (high number of false-negatives)
Management
Phlebotomy—1 unit every 2 to 4 weeks until hemoglobin (Hb) 11 g/dL, hematocrit (Hct) 35
Antimalarials (low dose)
Eliminate alcohol, estrogen, iron exposure
Sun protection/physical-block sunscreens
Referral to dermatologist, internist, gastroenterologist
Prognosis
Normal life span with clinical and biochemical remission achievable with treatment; premature death with hepatocellular carcinoma
Clinical Pearls
I refer most of my patients to a hepatologist for work-up … Urine fluorescence in the office is a fun test, but certainly not an adequate screen given high number of false-negatives … Plasma porphyrin screens are useful in following patients therapeutically but I have had difficulty recently with plasma porphyrin analysis from the major labs around the country, so I am sticking to urine for follow-up … Some studies have shown extremely high levels of hepatitis C in patients with porphyria cutanea tarda (PCT) … I get a hepatitis panel … I recommend titanium dioxide or mexoryl containing sunscreens … Women usually have electrolysis or use depilatories for the hypertrichosis … First line therapy is phlebotomy as an outpatient if there are no contraindications … Clinical response lags behind biochemical response … I’ve taken care of at least 30 patients, and I’ve never seen a failure with phlebotomy … There is no evidence suggesting that by simply having PCT leads to liver damage … I would want my residents to know how to differentiate VP from PCT… They can have identical cutaneous findings … Once you have a positive porphyrin screen, you must rule out a seizure history, and check the ratio of URO to COPRO in the urine… In PCT, 8:1; in VP, 1:1 or COPRO > URO. VD
|
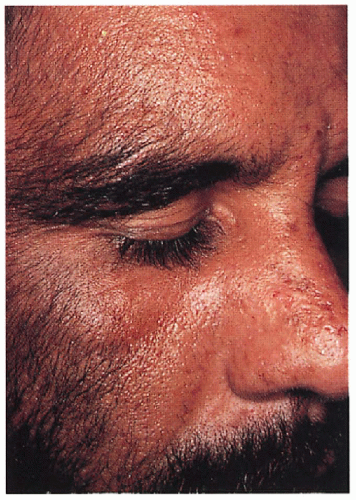 7.1. Facial hypertrichosis with erosions and scarring on nose. (5) |
 7.2. Urine porphyrin screen-coralpink fluorescence with Wood’s light. (5) |
Variegate Porphyria (VP)
Inheritance
Autosomal dominant; protoporphyrinogen (PROTOGEN) oxidase gene on 1q22; severe forms associated with hemochromatosis gene on 6p21
Prenatal Diagnosis
DNA analysis
Incidence
Most common in South Africa—1:330 in white population
Elsewhere approximately 1:50,000 to 100,000; M=F
Age at Presentation
Usually begins after puberty in second to third decade of life
Pathogenesis
Mutation in PROTOGEN oxidase gene causes a 50% decrease in PROTOGEN oxidase activity
Acute attacks precipitated by drugs (barbiturates, estrogen, griseofulvin, sulfonamides), infection, fever, alcohol, pregnancy, decreased caloric intake Increased Δ-aminolevulinic acid (ALA) synthetase with attacks
Key Features
Skin
Indentical to PCT with bullae, erosions, skin fragility, scarrring, millia, hypertrichosis, hyperpigmentation on photodistributed face, neck, and dorsum of hands
Acute Attacks (i.e., Acute Intermittent Porphyria and Hereditary Coproporphyra) Gastrointestinal
Colicky abdominal pain, nausea, vomiting, constipation
Central Nervous System
Peripheral neuropathy with pain, weakness, paralysis; confusional state, anxiety, depression, delerium, seizures, coma
Cardiovascular
Tachycardia, hypertension
Differential Diagnosis
Porphyria cutanea tarda (p. 216)
Acute intermittent porphyria (AIP; p. 220)
Hereditary coproporphyria (HCP; p. 221)
Laboratory Data
Plasma porphyrin level
Plasma porphyrin fluorescence spectrum—626 nm is diagnostic
Twenty-four-hour urine porphyrin levels—coproporphyrin = or > uroporphyrin
Urinary ALA and porphobillinogen (PBG) levels increased during attacks
Fecal porphyrin levels—markedly elevated; protoporphyrin > coproporphyrin
Management
Glucose loading, hematin infusion during attacks
Avoid drug precipitators, severe dieting
Referral to dermatologist—opaque sunscreens, topical antibiotics, β-carotene
Referral to neurologist—antiseizure medication, pain control
Referral to nutritionist—small carbohydrate meals to maintain glucose levels
Prognosis
Acute attacks may be life threatening and may leave residual neurologic damage
Clinical Pearls
Still most common in the Dutch Afrikaners of South Africa … 50% with combination skin and neurologic, 15% just neurologic, 35% just skin … Don’t bleed patients —you might rev up the cycle and lead to an attack … The trick is sun avoidance with titanium dioxide and mexoryl sunscreen … Need to work with neurologist who is aware of porphyria … Prior to surgery, the neurologist should talk with the anesthesiologist … Long periods of NPO (nothing by mouth) may be dangerous … Treat infection quickly … Hand patient “avoid these medications” list … Should they become pregnant?—tough question … Needs to be discussed with primary physician and obstetrician. VD
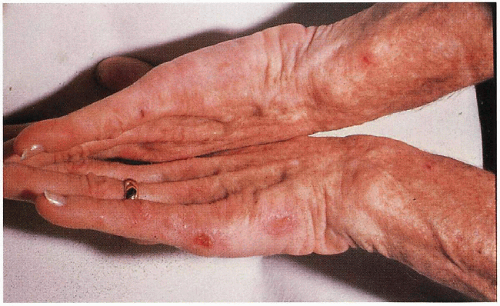 7.3. Bullae, erosions scattered on dorsum of hands in patient with variegate porphyria (VP). (5) |
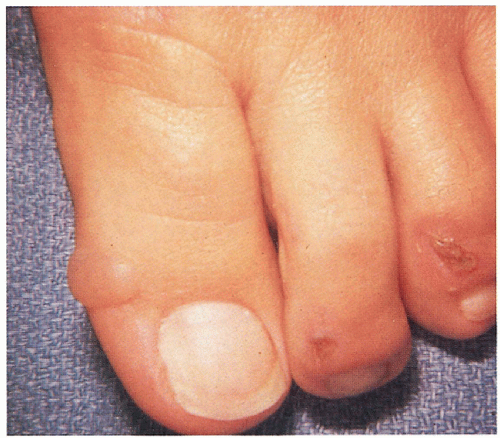 7.4. Patient with variegate porphyria (VP) with bullae, erosions on toes. (5) |
Acute Intermittent Porphyria (AIP)
Inheritance
Autosomal dominant; Porphobilinogen deaminase (PBGD) gene on 11q23
Prenatal Diagnosis
Amniocentesis: porphobilinogen (PBG) deaminase deficiency in cultured amniotic fluid cells
DNA analysis
Incidence
Approximately 1:66,000 worldwide; much higher in Scandinavia
M:F=1:1.5
Age at Presentation
Third to fourth decade of life
Pathogenesis
PBGD gene mutation causes a deficiency in PBGD activity
Acute attacks may be spontaneous or precipitated by drugs (see VP, p. 218), estrogen, infection, fever, decreased caloric intake, alcohol, pregnancy, menses
Increased ALA synthetase with attacks
Mechanism for attacks unknown
Key Features
Acute Attacks
Central Nervous System
Peripheral neuropathy with pain, weakness, paralysis; anxiety, depression, seizures, confusional state, delirium, coma; rarely, respiratory failure caused by paralysis
Gastrointestinal
Colicky abdominal pain, nausea, vomiting, constipation
Cardiovascular
Tachycardia, hypertension
Hematologic
Hyponatremia secondary to antidiuretic hormone (ADH) secretion
Differential Diagnosis
VP (p. 218)
Hereditary coproporphyria (p. 221)
Acute abdomen
Organic neurologic/psychiatric disease
Laboratory Data
Enzyme assay—decreased PBGD in red blood cells
Plasma porphyrin level and fluorescence spectrum
Twenty-four-hour or spot urine—increased ALA and porphobilinogen during and between attacks; dark, port wine-colored urine
CBC with differential (leukocytosis), chemical screen
Management
Glucose load, hematin infusion during acute attacks
Referral to neurologist—antiseizure medication, pain control
Avoid precipitators
Referral to nutritionist—small carbohydrate meals to maintain glucose levels
Fluid and electrolytes monitored
Check family for carrier status with enzyme assay
Prognosis
Improved with avoidance of precipitators; acute attacks may be life threatening and leave residual neurologic deficits
Clinical Pearls
I can see these patients being labeled as having “chronic fatigue syndrome” … A lot of people end up having multiple exploratory laporatomies … They’ve seen multiple physicians including gastrointestinal, neurologic, and psychiatric … Many are thought to be crazy … Always give list of medicatoins to avoid … Pregnancy, surgical issues must be discussed with patients and their physicians … They can die from their attacks … But I don’t think it’s a high mortality rate at all … Like other seizure patients, they shouldn’t swim alone. VD
Hereditary Coproporphyria (HCP)
Inheritance
Autosomal dominant; coproporphyrinogen (COPROGEN) oxidase gene on 3q12
Prenatal Diagnosis
DNA analysis
Incidence
Rare—estimated 2 per million in Denmark; M=F; symptomatic M:F=1:2.5
Age at Presentation
Third to fourth decade of life
Pathogenesis
COPROGEN oxidase gene mutation causes the phenotype
Acute attacks precipitated by same factors as AIP and VP
Increased ALA synthetase with attacks
Key Features
Skin (approximately 30% of symptomatic patients)
Laboratory Data
Plasma porphyrin level and fluorescence spectrum
Increased coproporphyrin levels in stool and urine
Increased ALA and PBG in urine during attacks only
Prognosis
Acute attacks may be life threatening; many gene carriers are asymptomatic throughout life
Clinical Pearls
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


