Disorders of Pigmentation
Seth Orlow M.D.
Amy Paller M.D.
Jean Bolognia M.D.
D.G.R. Evans M.D.
Clinical Pearls
(SO)
(AP)
(JB)
(DE)
Oculocutaneous Albinism Type 1 (OCA1)
Synonym
Tyrosinase-negative albinism
Inheritance
Autosomal recessive; tyrosinase (TYR) gene on 11q14-q21
Prenatal Diagnosis
DNA mutation analysis
Incidence
1:28,000 blacks; 1:39,000 caucasians; M=F
Age at Presentation
Birth
Pathogenesis
Mutations in the TYR gene lead to absent tyrosinase activity or lack of tyrosinase transport to melanosomes
Normal number of melanocytes
Unable to produce melanin in skin, hair and eyes
Only stage I and II premelanosomes in melanocytes
Miswiring of optic fibers
Key Features
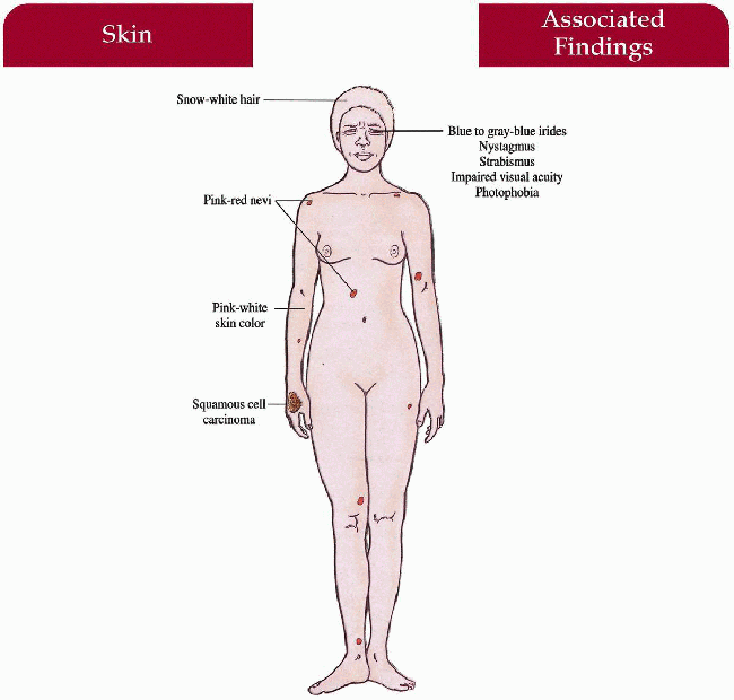
Skin
Generalized pink-white color, solar keratoses, pink-red nevi, squamous cell cancers > basal cell cancers > melanoma
Hair
Snow-white color
Eyes
Blue to gray-blue irides, severe nystagmus, photophobia, impaired visual acuity (20/200 or worse), prominent red reflex throughout life, strabismus, foveal hypoplasia
Differential Diagnosis
OCA1B (yellow mutant, minimal pigment, and temperature-sensitive albinism)
OCA2 (p. 58)
Hermansky-Pudlak syndrome (p. 60)
Chédiak-Higashi syndrome (p. 62)
Prognosis
Skin and hair do not improve with age
Vision remains stable or worsens with age
Clinical Pearls
Almost certain to get squamous cell cancer, just a matter of when … Some may need to be placed in special classes for the visually impaired … Referral to NOAH support group (see Support Groups). SO
See OCA2 (p. 58)
|
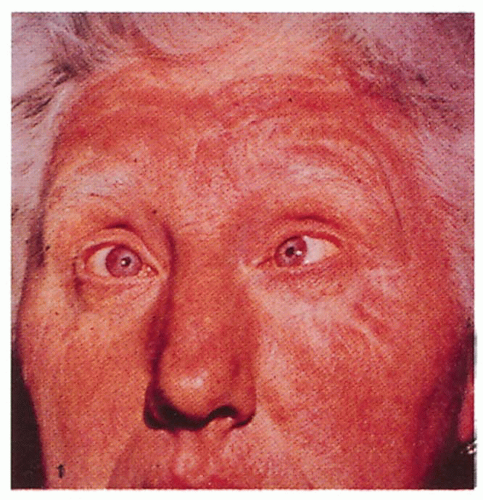 2.1. Snow-white hair, pink skin, strabismus, and gray-blue irides in an adult. (24) |
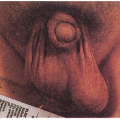
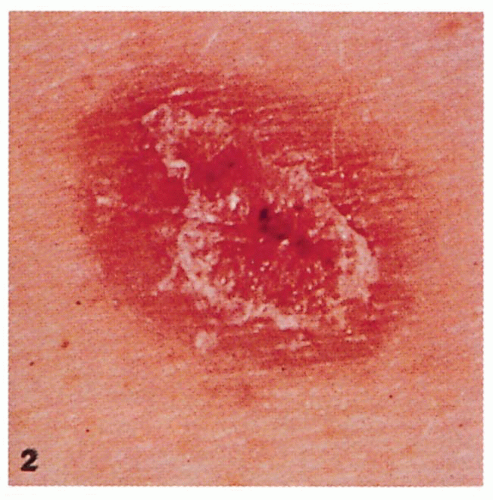 2.2. Malignant melanoma presenting as a pink macule with crust. (24) |
Oculocutaneous Albinism Type II (OCA2)
Synonym
Tyrosinase-positive albinism
Inheritance
Autosomal recessive; P gene on 15q11.2-12
Prenatal Diagnosis
DNA linkage analysis and mutation detection available
Incidence
1:15,000 blacks; 1:37,000 whites; M=F
Most common form of albinism
Age at Presentation
Birth
Pathogenesis
Mutation in P gene with decrease eumelanin synthesis; gene product thought to play a role in tyrosinase transport
(+) Tyrosinase, normal number of melanocytes
Decreased melanin in skin, hair, and eyes
Miswiring of optic fibers
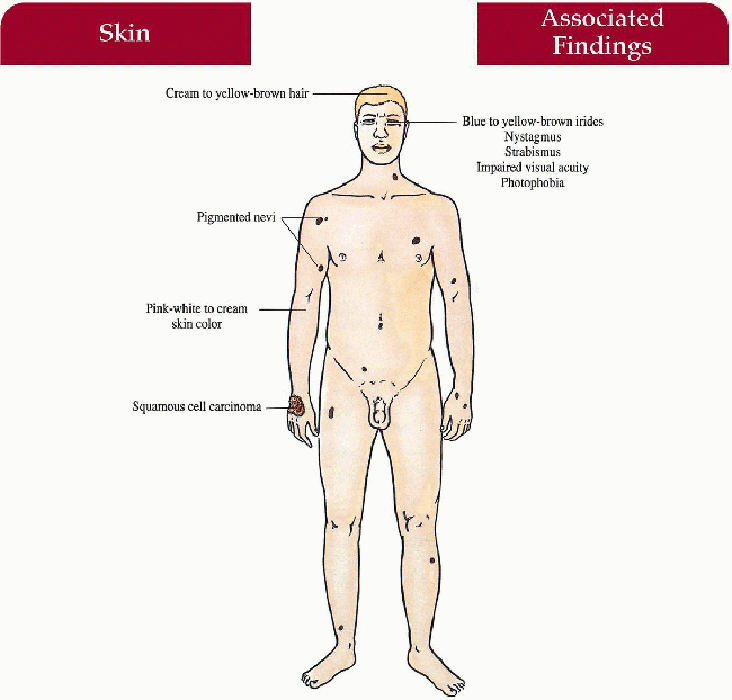
Key Features
Skin
Generalized pink-white to cream color
Multiple pigmented nevi, ephelides, and lentigines increase with age
Solar keratoses, squamous cell and basal cell cancers with age
Hair
Cream to yellow-brown color
Eyes
Blue to yellow-brown irides (race dependent), nystagmus, photophobia, impaired visual acuity, strabismus, foveal hypoplasia
Differential Diagnosis
OCA1, OCA1B (p. 56)
Hermansky-Pudlak syndrome (p. 60)
Chédiak-Higashi syndrome (p. 62)
Laboratory Data
DNA mutation analysis
Management
Skin
Sun avoidance (especially mid-day): broad-spectrum sunscreen, long-sleeved shirts, brim hat
Referral to dermatologist—skin cancer screening every 6 months
Eyes
UVB-blocking sunglasses, corrective lenses, tinted glasses/contact lenses
Referral to ophthalmologist
Prognosis
Skin and hair pigment increases over time; eye symptomatology may improve with age
Clinical Pearls
Skin
More difficult problem for darker races with increased social ostracism (less than with vitiligo, however) … Referral to NOAH support group (see Support Groups).
Eyes
Most difficult problem for caucasians but can be equally bad for blacks depending on the level of nystagmus and severity of albinism. SO
|
 2.3. Yellow-brown hair and pink skin in a black girl. (5) |
 2.4. Melanin production after hair bulb incubation test. (1) |
Hermansky-Pudlak Syndrome
Inheritance
Autosomal recessive; gene loci HPS1 gene on 10q23 (most common, found in northwest Puerto Rico and a small village in Swiss Alps), AP3B1 gene on 5q14.1 (HPS2); there are five other gene loci identified to date
Prenatal Diagnosis
DNA mutation analysis if defect known
Incidence
Over 200 case reports; increased frequency in Puerto Rico, Holland
Age at Presentation
Birth
Pathogenesis
Mutation in HPS1 gene is a 16-bp duplication of the gene important in protein localization via intracellular trafficking and organelle formation
AP3B1 gene mutations cause HPS2—the gene encodes the β-3a subunit of AP3 adapter complex, which is thought to be important in protein packaging, vesicular formation, and membrane fusion within the cell
Tyrosinase-positive
Bleeding diathesis secondary to platelet storage pool defect with decreased adenosine diphosphate (ADP), adenosine triphosphate (ATP), and serotonin granules—impairs platelet aggregation
Lysosomal membrane defect—accumulation of ceroid lipofuscin in macrophages within the lung and gastrointestinal tract
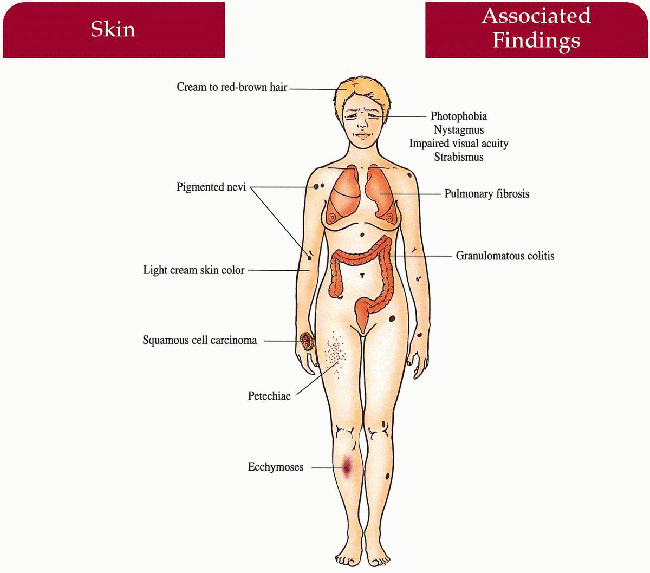
Key Features
Skin
Pigment dilution dependent on race
Pigmented nevi, solar keratoses, squamous cell cancer, basal cell cancer
Ecchymoses, petechiae
Hair
Cream to red-brown color
Eyes
Photophobia, nystagmus, decreased visual acuity, strabismus, foveal hypolasia
Hematologic
Epistaxis, gingival bleeding, menorrhagia, prolonged bleeding during childbirth, dental extraction, surgery
Lymphohistiocytic
Ceroid (chromolipid) deposition in macrophages in the lung (pulmonary fibrosis), gastrointestinal tract (granulomatous colitis), cardiac muscle (cardiomyopathy)
Laboratory Data
Bleeding time prolonged; prothrombin time/partial thromboplastin time (PT/PTT), platelet count normal
Wet-mount electron microscopy—demonstrates platelets without dense granules
Management
Avoid aspirin and other prostaglandin synthesis inhibitors
Baseline chest x-ray at early age
Pulmonary function test and colonoscopy if symptomatic
Solar protection and avoidance
Referral to dermatologist, hematologist-oncologist, ophthalmologist, and symptom-specific subspecialist
Educate dentist, obstetrician, and surgeon prior to dental extraction, delivery, and surgical procedure
Prognosis
Premature death secondary to hemorrhage, colitis, pulmonic disease, squamous cell cancer; otherwise normal life span
Clinical Pearls
Must first establish patient is a tyrosinase positive albino … Elicit country of origin; remember large Puerto Rican population in the northeast United States … Never give aspirin … Dental surgery and minor surgery may need to be done in hospital setting with platelets available … Approximately 33% of patients will develop symptomatic pulmonary fibrosis. SO
|
 2.5. Female with showers of petechiae and creamcolored hair and skin. (25) |
 2.6 Top: Normal platelets with dense granules. Bottom: Platelets from Hermansky-Pudlak patient with absent granules. (26) |
Chédiak-Higashi Syndrome
Inheritance
Autosomal recessive; LYST gene on 1q42; high consanguinity
Prenatal Diagnosis
Fetoscopy—fetal hair shafts reveal clumping of melanosomes on light microscopy; fetal blood reveals characteristic neutrophilic granules
Incidence
Rare; fewer than 100 cases reported; M=F
Age at Presentation
Birth to a few months old
Pathogenesis
LYST gene mutation codes for a lysosomal tracking protein that regulates microtubule-mediated lysosomal fusion/fission and protein sorting; this defect leads to accumulation of giant lysosomal granules in a variety of cells:
Neutrophils—defective phagocytosis, decreased chemotaxis
Melanocytes—pigmentary dilution
Neurons—progressive neurologic deterioration
Platelet storage pool deficiency—bleeding diathesis
Decreased natural killer cell and antibody-dependent cell-mediated cytolysis function
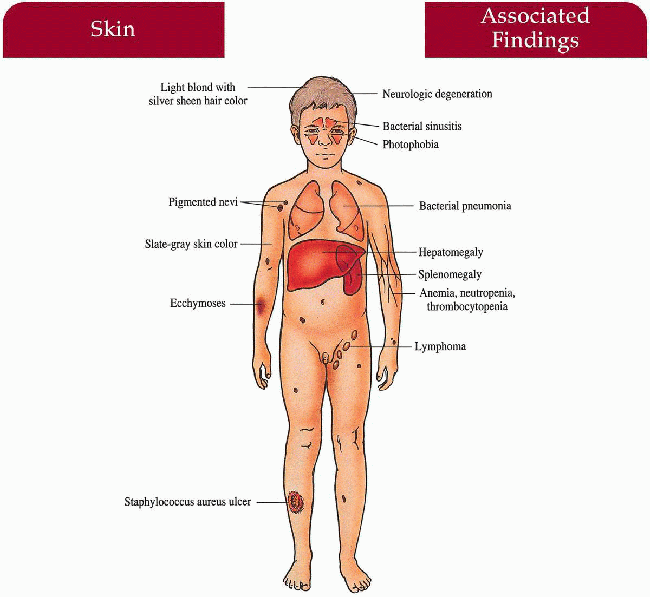
Key Features
Skin
Light cream to slate-gray color, recurrent bacterial infections—Staphylococcus aureus most common
Hair
Light blonde color (in caucasians) with a silver sheen
Eyes
Photophobia, strabismus, nystagmus, decreased uveal pigment
Upper and Lower Respiratory Tract
Recurrent bacterial sinusitis, pneumonia
Central Nervous System
Progressive neurologic deterioration with ataxia, muscle weakness, sensory loss, seizures (rare)
Accelerated Phase (approximately 85% of patients):
Hematologic
Lymphohistiocytic proliferation with infiltration of liver, spleen, and lymph nodes; associated anemia, neutropenia, thrombocytopenia manifested as petechiae, ecchymoses, gingival bleeding, epistaxis, gastrointestinal hemorrhage, overwhelming infection
Differential Diagnosis
OCA2 (p. 58)
Hermansky-Pudlak syndrome (p. 60)
Chronic granulomatous disease (p. 258)
Griscelli syndrome (p. 64)
Elejalde syndrome
Laboratory Data
Diagnostic complete blood cell count (CBC) revealing giant granules in neutrophils
Management
Referral to pediatric hematologist-oncologist-bone—marrow transplant, chemotherapy, high-dose ascorbic acid
Antibiotics, sun protection
Referral to dermatologist, infectious disease specialist, immunologist, neurologist, and ophthalmologist
Prognosis
Death by late childhood from overwhelming infection or hemorrhage during lymphoma-like phase; may reverse deterioration with bone marrow transplantation
Clinical Pearls
Bone marrow transplant is the treatment of choice and should be performed early on before the accelerated phase takes place … Lymphoma-like phase responds poorly to chemotherapy … Platelet storage pool defect less severe than in Hermansky-Pudlak … Death usually by 10 years of age. SO
|
 2.7. Black child with silver-gray hair and skin. (27) |
 2.8. Giant lysosomal granules in a polymorphonuclear neutrophil. (27) |
Griscelli Syndrome
Inheritance
Autosomal recessive
Prenatal Diagnosis
Fetal scalp biopsy at 21 weeks’ gestation: hair evaluation; fetal blood sample: leukocyte evaluation
DNA analysis
Incidence
Rare—less than 40 cases reported; M=F
Age at Presentation
First year of life
Pathogenesis
Mutations in gene encoding for myosin Va or RAB27a, proteins involved in organelle trafficking and membrane transport; melanophilin gene mutations also implicated in subset of patients
Key Features
Skin
Pigmentary dilution; cutaneous pyogenic infections, abscesses
Hair
Silver-gray hair, eyebrows, eyelashes
Hematologic
Neutropenia, thrombocytopenia, without leukocyte inclusions
Immunologic
Lymphohistiocytic infiltration leading to hepatosplenomegaly, combined T- and B-cell immunodeficiency; accelerated lymphoma-like phase (i.e., Chediak-Higashi) often occurs
Infectious Disease
Episodic fever with/without infection, pyogenic systemic infections
Neurologic
Progressive deterioration with hypotonia, psychomotor retardation, seizures
Differential Diagnosis
Chédiak-Higashi syndrome (p. 62)
Elejalde syndrome
Chronic granulomatous disease (p. 258)
OCA2 (p. 58)
Laboratory Data
Hair—uneven clumps of melanin in medulla of hair shaft on light microscopy
Complete blood count (CBC)—absent cytoplasmic inclusion bodies in neutrophils
Management
Referral to hematology-oncology-bone marrow transplant
Referral to infectious disease
Referral to neurologist
Referral to dermatology-assist in diagnosis
Prognosis
Progressive deterioration may be aborted with bone marrow transplantation
Clinical Pearls
Patients with Griscelli syndrome caused by mutations in the MYO5A gene develop primary neurologic impairment (e.g., severe developmental delay) but not hemophagocytic syndrome (a phenotype similar to dilute mice] whereas those with RAB27A mutations develop a hemophagocytic syndrome and any neurologic involvement is secondary, i.e., caused by lymphocytic infiltration of the central nervous system (CNS), which is a phenotype similar to ashen mice. JB
|
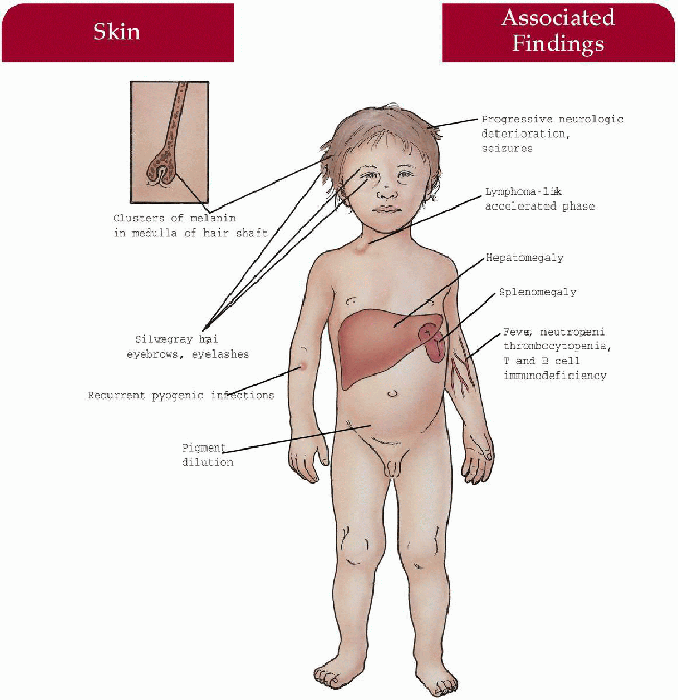
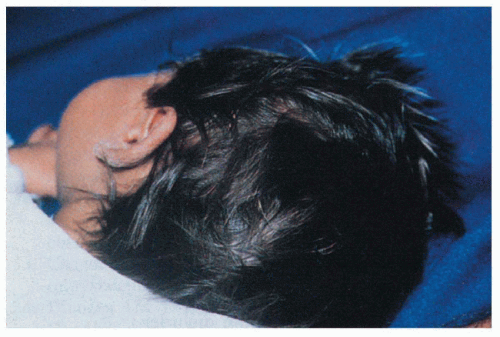 2.9. Hair with metallic silvery-gray sheen. (28) |
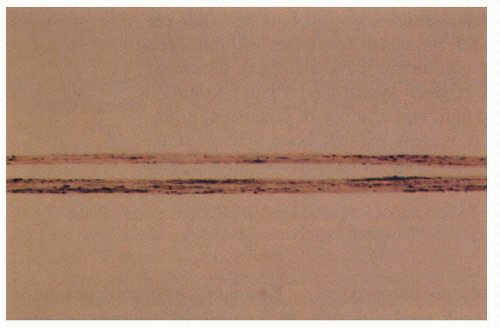 2.10. Light microscopy of hair reveals irregular, large clumps of melanin pigment. (28). |
Piebaldism
Synonym
Familial white spotting
Inheritance
Autosomal dominant; mutation in c-kit proto-oncogene on chromosome 4q12
Prenatal Diagnosis
DNA linkage analysis and mutation detection available
Incidence
Less than 1:20,000; all races; M=F
Age at Presentation
Birth
Pathogenesis
A mutation in the c-kit proto-oncogene results in abnormal tyrosine kinase transmembrane receptors, decreases signal transduction, and causes abnormal melanocyte embryogenesis with defective melanoblast proliferation, migration and distribution.
Key Features
Skin
Depigmented patches on mid-forehead, central eyebrows, neck, anterior trunk, mid-extremities; often bilateral, sparing the hands, feet, back, shoulders, hips Islands of hyperpigmented to normally pigmented patches within and at the borders of hypopigmentation
Hair
White forelock (80% to 90%)
Rare Case Reports
Hirschsprung disease, mental retardation, deafness, cerebellar ataxia
Laboratory Data
Histology from depigmented area reveals decreased number or absent melanocytes and melanin
Management
Autologous cultured melanocyte grafts
Sunscreen
Camouflage—hair dye, Dermablend; 20% topical monobenzyl ether of hydroquinone
Prognosis
Pigmentary alteration usually stable and permanent; normal life span
Clinical Pearls
Melanocyte transplant technology not perfected yet … Coping mechanisms seem to be better for patients with a congenital skin defect (piebaldism) rather than an acquired skin defect (vitiligo). SO
|
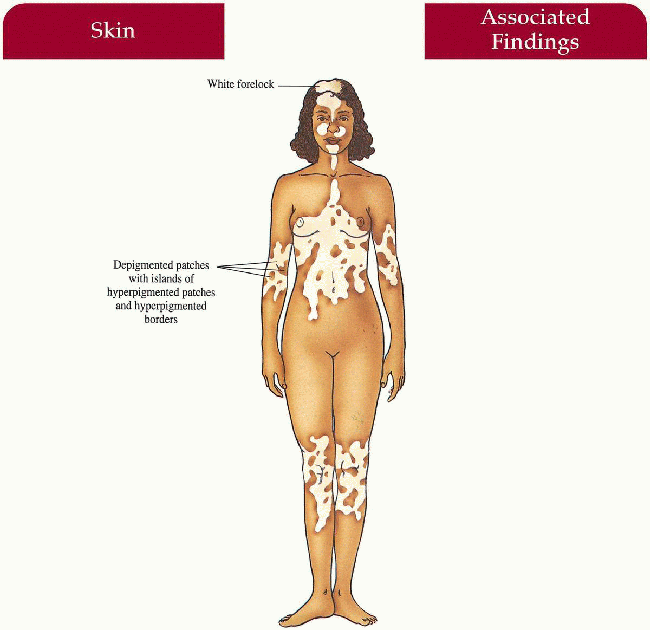
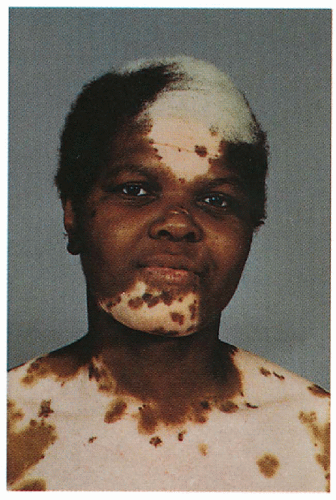 2.11. Black female with white forelock and depigmented patches with islands of hyperpigmentation on her mid-face and central trunk. (29) |
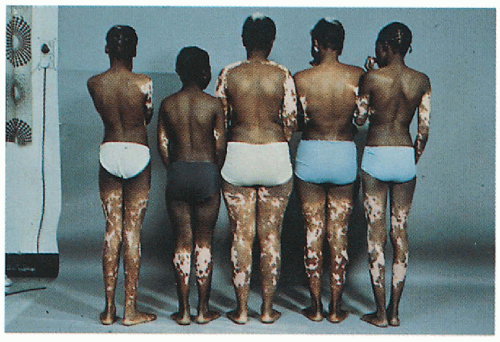 2.12. Family of patient in Figure 2.11 with similar cutaneous findings. (29) |
Waardenburg Syndrome
Inheritance
Autosomal dominant; Pax3 gene on 2q35 in I and III, MITF gene on 13q in II, and the SOX1O and endothelin-3 genes on 22q13 and 20q13 respectively in IV
Prenatal Diagnosis
DNA analysis if gene defect known
Incidence
1:42,000; M=F; all races; 1% to 3% of all congenitally deaf children
Age at Presentation
Birth
Pathogenesis
Four distinct types have been defined by their unique clinical and genetic features: Types I and III are caused by mutations in PAX3, a transcription factor that controls neural crest differentiation and regulates transcription of other genes downstream including those responsible for melanoblast activation and migration, inner ear structures and facial bony and cartilaginous structures.
Type II is caused by a mutation in the MITF gene, the melanocyte transcription factor.
Three genetic etiologies involving control of neural crest development have been described for producing the phenotype seen in type IV: SOX1O transcription factor mutations, endothelin-3 signaling gene mutations and endothelin receptor gene mutations.
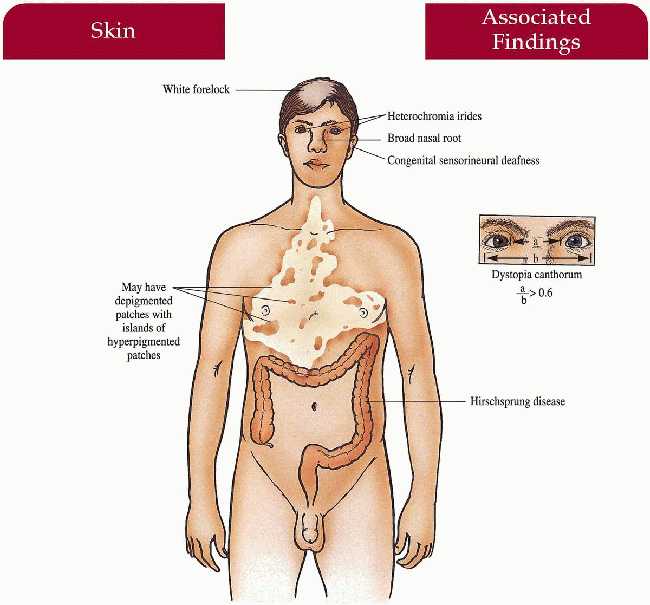
Key Features
Skin
May have depigmented patches on body
Hair
White forelock (< 50%), synophrys (70%)
Teeth
Caries
Nose
Broad nasal root (80%)
Eyes
Dystopia canthorum (99% but not in II)—lateral displacement of medial canthi with normal interpupillary distance, complete or partial heterochromia irides (25%), hypopigmented fundus
Ears
Congenital sensorineural hearing loss (20%, most common in II)
Colon
Hirschsprung disease (< 5%, exclusively IV)
Musculoskeletal
Cleft lip/palate (I), upper limb, and pectoral anomalies (III)
Differential Diagnosis
Piebaldism (p. 66)
Vogt-Koyanagi-Harada syndrome
Vitiligo
Alezzandrini syndrome
Laboratory Data
Management
Referral to audiologist, otolaryngologist, ophthalmologist
Referral to gastroenterologist and surgeon if symptomatic
Prognosis
Heterochromia irides and white forelock may fade after 1 year; normal life span
Clinical Pearls
Most important, all patients must see audiologist early on to impact upon learning and phonation in the congenitally deaf … Benefit from hearing aids … Colored contacts useful for heterochromia irides … Watch for gastrointestinal symptomatology in newborn/infant … Elicit bowel habit history … Dermablend, hair dyes useful. SO
|
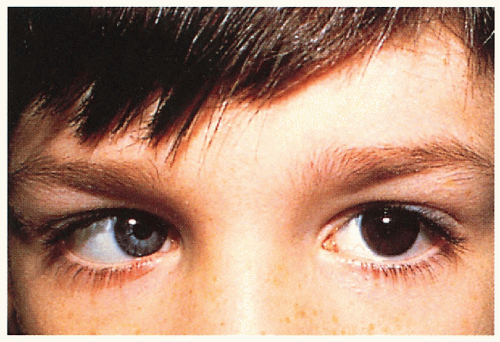 2.13. Boy with heterochromia irides, strabismus, broad nasal root, and dystopia canthorum. (1) |
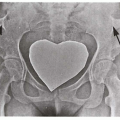
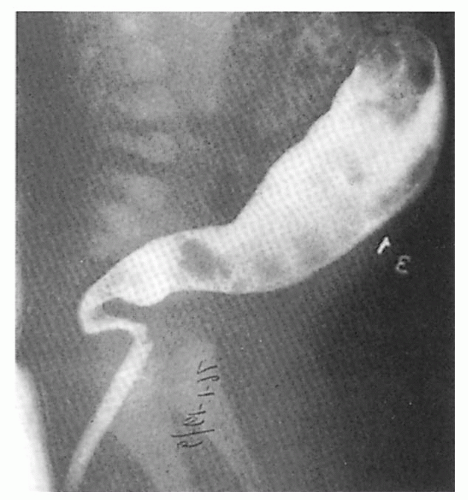 2.14. X-ray depicting tapering of normal colon in patient with Hirschsprung disease. (30) |
Hypomelanosis of Ito
Synonym
Incontinentia pigmenti achromians
Inheritance
Not inherited; chromosomal or single gene mosaicism
Prenatal Diagnosis
None
Incidence
Rare; all races; M=F
Age at Presentation
Birth to 1 year old
Pathogenesis
The cutaneous phenotype reflects many different forms of genomic mosaicism
Key Features
Skin
Unilateral and bilateral whirled marble cake hypopigmentation in Blaschko’s lines
Hair
Alopecia
Associated Findings (seen in 75% of cases):
Central Nervous System
Seizures, mental and motor retardation
Eyes
Strabismus, hypertelorism
Musculoskeletal
Scoliosis, limb length discrepancy
Teeth
Anodontia, dental dysplasia
Differential Diagnosis
Nevus depigmentosus
Tuberous sclerosis (p. 88)
Incontinentia pigmenti (p. 72)
Segmental vitiligo
Laboratory Data
None
Management
Complete physical examination by primary care physician
Referral to subspecialist if symptomatic
Camouflage cosmetics
Prognosis
Hypopigmentation may fade with time; normal life span
Clinical Pearls
Hypopigmentation is not a static finding … I have seen patients revert to normal pigmentation … Nevus depigmentosus, hypomelanosis of Ito, and linear and whorled nevoid hypermelanosis represent a spectrum of phenotype related to various mosaic genotypes. SO
|
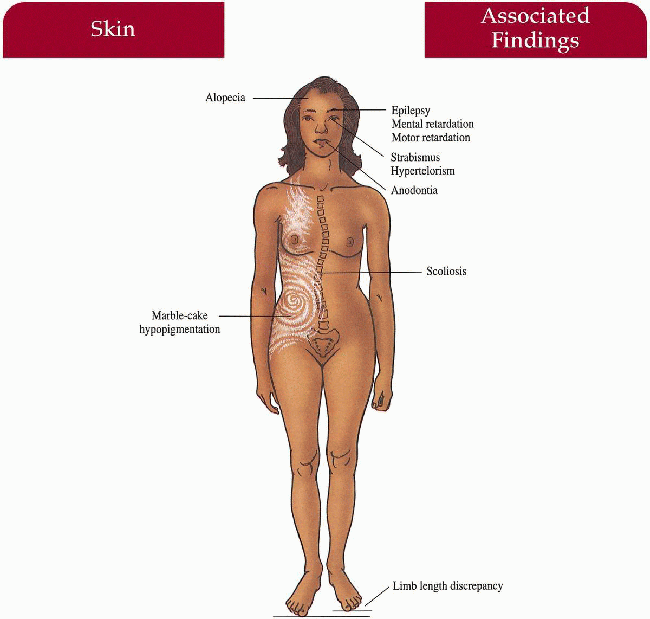
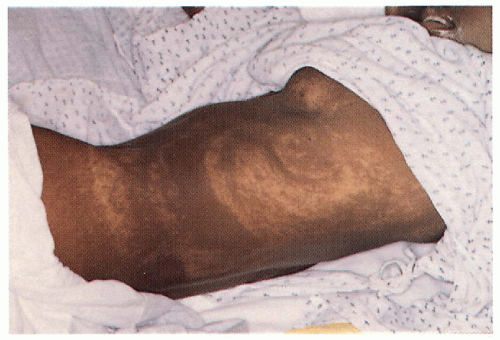 2.15. Eighteen year-old girl with marble-cake hypopigmentation who suffered from intractable seizures, mental and motor retardation. |
 2.16. Same patient with anodontia. |
Incontinentia Pigmenti
Synonym
Bloch-Sulzberger syndrome
Inheritance
X-linked dominant; rare male survivors thought to have Klinefelter syndrome; NEMO gene on Xq28
Prenatal Diagnosis
DNA analysis if gene known in family
Incidence
Over 700 cases reported; 97% female
Age at Presentation
Birth to first few weeks of life
Pathogenesis
Mutation in NEMO (NF-κB essential modulator) gene leads to defective NF-κB activation (80% have identical mutation secondary to gene rearrangement in paternal meiosis). NF-κB is a transcription factor essential for several inflammatory, immune and apoptotic pathways
Key Features
Skin
Stage I
Vesicular (birth to 1 to 2 weeks): vesicles and bullae in a linear arrangement on extremities, trunk, and scalp; erythematous macules and papules
Stage II
Verrucous (2 to 6 weeks): streaks of hyperkeratotic papules, pustules, and papules on extremities
Stage III
Hyperpigmentation (3 to 6 months): whorls and swirls of hyperpigmentation along Blaschko’s lines
Stage IV
Hypopigmentation (second to third decade): hypopigmented whorls and swirls replacing hyperpigmentation; with/without follicular atrophy
Hair
Scarring alopecia (30%)
Nails
Dystrophic changes (5% to 10%)
Teeth
Anodontia, peg/conical teeth (66%); deciduous and permanent affected
Eyes
(25% to 35%)
Strabismus, cataracts, optic atrophy, retinal vascular changes with secondary blindness, retrolental mass
Central Nervous System
(30%)
Seizures, mental retardation, spastic paralysis
Differential Diagnosis
Neonate
Epidermolysis bullosa (p. 200)
Impetigo
Herpes simplex virus
Epidermolytic hyperkeratosis (p. 6)
Congenital syphilis
Laboratory Data
Skin biopsy in vesicular stage—abundant eosinophils
Complete blood count—peripheral eosinophilia in infancy
Management
Referral to dermatologist—diagnosis, topical care
Referral to dentist at 1 year old
Referral to ophthalmologist at time of diagnosis
Referral to neurologist if symptomatic
Prognosis
Normal life span
Clinical Pearls
Dentist can provide dentures, prosthodontics for correction of dysfunctional teeth … Refer to ophthalmologist to fix strabismus, cataracts, and retrolentalfibroplasia-like ocular disease … I let the clinical exam dictate my work-up of other systems … Genetic counseling extremely important … Affected women may have a very difficult time conceiving and an increased rate of miscarriages … Always examine Mom. SO
 2.17. Bullae and verrucous papules along Blaschko’s lines on infant’s trunk and extremity. (4) |
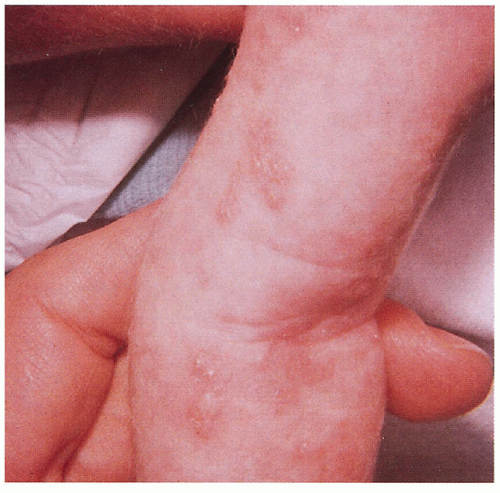 2.18. Close-up of bullae in a linear distribution on infant’s arm. (1) |
 2.19. Whorls and swirls of hyperpigmentation in Blaschko’s lines. (1)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|