Disorders of Metabolism
Kurt Hirschhorn M.D.
Judith Willner M.D.
Clinical Pearls
(KH)
(JW)
Alkaptonuria
Synonym
Ochronosis
Inheritance
Autosomal recessive; homogentisate 1,2-dioxygenase (HGO) gene on 3q21-q23
Prenatal Diagnosis
DNA analysis
Incidence
1:250,000; increased in Dominican Republic, Slovakia; M=F
Age at Presentation
Childhood (dark cerumen, black-stained underwear) to adulthood (skin pigment, arthropathy)
Pathogenesis
Mutations in the HGO gene leads to deficiency of homogentisic acid oxidase with secondary accumulation of homogentisic acid in connective tissue
Key Features
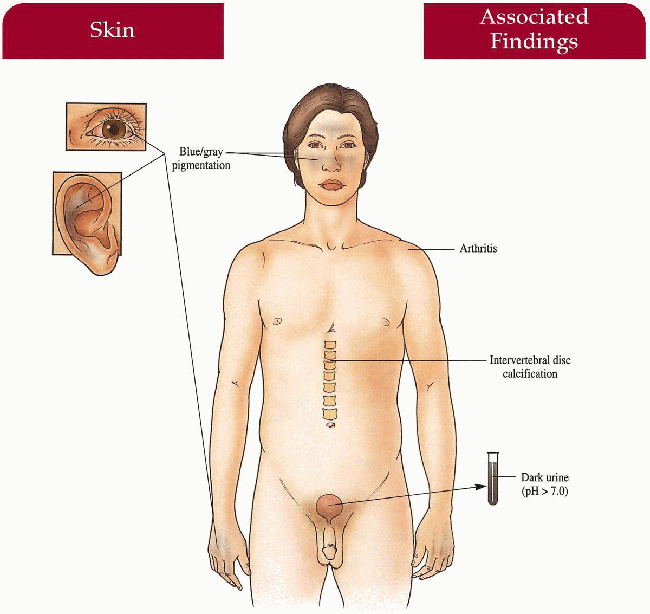
Skin
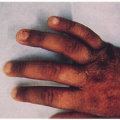
Blue-gray pigmentation increased on nose, cheeks, forehead, axillae; blue-gray pigmented cartilage and tendons visualized through skin on ears, nose tip, extensor hands, costochondral junctions; brown/black cerumen, sweat
Eyes
Blue-gray scleral pigment
Musculoskeletal
Severe arthropathy involving larger joints including hip, knee, shoulder, spine; intervertebral disc calcification
Genitourinary
Dark urine with pH above 7.0 (diapers, clothing discolored after cleansing with alkaline soaps)
Cardiovascular
Mitral and aortic valvulitis; increased incidence of myocardial infarction later in life
Differential Diagnosis
Exogenous ochronosis (antimalarials, hydroquinone)
Argyria
Chrysoderma
Amiodarone administration
Laboratory Data
Enzyme assay: measurement of urinary homogentisic acid
Darkening of urine with addition of NaOH
Spine films
Electrocardiogram (ECG) in older patients
Management
Analgesics, physical therapy, joint replacement for arthropathy
Supplemental vitamin C up to 1 g per day for older children and adults
Reduction of phenylalanine and tyrosine may help reduce homogentisate excretion
Prognosis
Normal life span with persistent pigmentation changes and unremitting arthropathy; older patients with increased incidence of myocardial infarction
Clinical Pearls
With advent of disposable diapers, noticing dark-stained cloth diapers after washing no longer helps make the diagnosis… Most of the time, parents/patients never notice black urine… Degenerative surfaces and narrowing of joint space on x-ray… Historically, this syndrome is very important because it is the first biochemical disease described by Garrod… He is the man who invented biochemical genetics, termed the words “inborn errors of metabolism,” and came up with the idea that we’re all biochemically different… Interestingly, the diagnosis has been made in Egyptian mummies with black cartilage. KH, JW
|
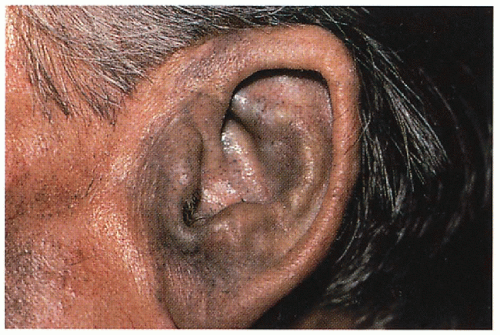 11.1. Blue-gray pigmentation involving ear cartilage. (120) |
 11.2. Similar pigmentation on patient’s hands. (120) |
Fabry Disease
Synonym
Angiokeratoma corporis diffusum
Inheritance
X-linked recessive; α-galactosidase A (GLA) gene on Xq21.33-q22
Prenatal Diagnosis
Chorionic villus sampling (CVS)/amniocentesis—α-galactosidase A enzyme assay DNA analysis
Incidence
Approximately 1:40,000 males; female heterozygotes reported with marked variability in expression
Age at Presentation
Childhood to adolescence
Pathogenesis
Mutation in GLA leads to defective activity of α-galactosidase A and accumulation of neutral glycosphingolipids with preferential deposition in vascular endothelium resulting in ischemia and infarction; also deposits within most tissues of the body, including heart and kidney
Key Features
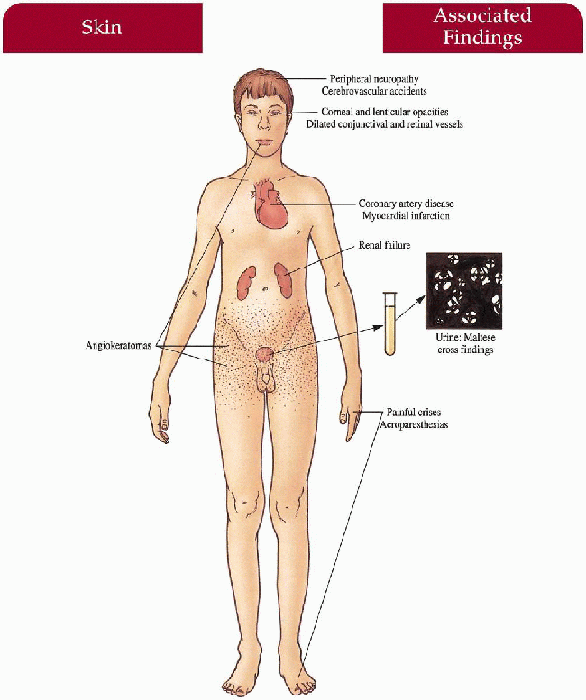
Skin
Angiokeratomas—dark red to blue-black papules with/without overlying hyperkeratosis concentrated symmetrically between the umbilicus and knees, increase in number and size with age; hypoanhidrosis
Mucous Membranes
Angiokeratomas—oral mucosa, conjunctiva
Peripheral Nervous System
Painful crises—most severe on hands and feet but can spread proximally; exercise, fever, climate/temperature changes, emotional stress may trigger episode Acroparesthesias—constant discomfort of hands and feet with burning, tingling paresthesias
Cardiovascular
Angina, myocardial infarction, conduction defects, mitral insufficiency
Kidney
Progressive renal deterioration with proteinuria, birefringent lipid globules (“maltese crosses”) seen with polarizing microscopy, renal failure
Central Nervous System
Peripheral neuropathy, cerebrovascular accidents
Eyes
Characteristic corneal opacities with whorl-like configuration, lenticular opacities, dilated and tortuous conjunctival and retinal vessels
Differential Diagnosis
Rheumatic fever
Mercury/heavy metal poisoning
Erythromelalgia
Other angiokeratomas: angiokeratoma of Fordyce, fucosidosis, sialidosis, β-galactosidase deficiency, aspartylglucosaminuria
Laboratory Data
DNA analysis
Enzyme assay—deficient α-galactosidase A activity
Skin, bone marrow biopsy
Urinary sediment examination with polarizing microscopy
Slit-lamp ophthalmologic examination
ECG
Management
α-Galactosidase A intravenous replacement therapy
Diphenylhydantoin, carbamazepine—pain crises
Symptomatic care of cardiac, central nervous system (CNS), and ocular manifestations
Long-term hemodialysis, renal transplantation
Advise physical education teachers/occupational advice—minimize physical/emotional stresses
Prognosis
Premature death during fifth decade secondary to myocardial infarction, cerebrovascular accidents, and renal failure; enzyme replacement therapy, hemodialysis, renal transplantation may extend life span
Clinical Pearls
Acral pain and paresthesias are very specific findings… You virtually never see them except in mercury/heavy metal poisoning… Pain is so severe that patients develop very peculiar behavior to help deal with it… Such as dunking their hands and feet in cold toilet water… The kid is often labeled “nuts” by parents and medical community… Low-dose dilantin works very well for the pain… Preliminary studies seem to show a reduction in the accumulation of glycosphingolipids… Interestingly, some transplanted patients show improvement in their pain… Group B blood groups tend to do worse… Of all X-linked recessive diseases, this one has the largest number of symptomatic female carriers… Enzyme replacement therapy is now available. KH, JW
 11.3. Angiokeratomas on penile shaft, scrotum, groin, and inner thigh. (121) |
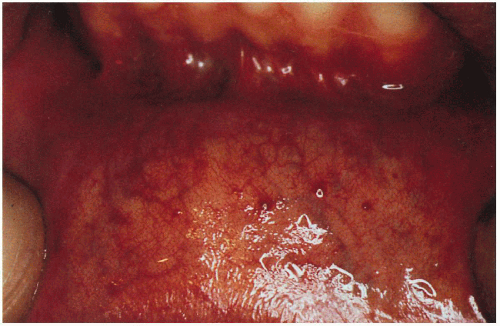 11.4. Angiokeratomas studding the labial mucosa. (122) |
|
Gaucher Disease
Inheritance
Autosomal recessive; acid-β-glucocidase (GBA) gene locus 1q21
Prenatal Diagnosis
CVS/amniocentesis—glucocerebrosidase enzyme assay
Ultrasound: hydrops fetalis, hepatosplenomegaly may be seen in type II disease
DNA analysis
Incidence
Over 350 patients reported; type I is 20 times more common than type II and increased in Ashkenazi Jewish population; M=F
Age at Presentation
Type II (infantile)—2 to 3 months of life
Type I (adult)—may begin within first decade of life; usually begins later in adulthood
Pathogenesis
Mutation in GBA gene leads to decreased glucocerebrosidase activity resulting in accumulation of glucocerebroside in histiocytes (Gaucher’s cells) in spleen, liver, bone marrow, lymph nodes, and brain (infantile only); adult form without CNS accumulation secondary to adequate neuronal glucocerebrosidase activity
Key Features
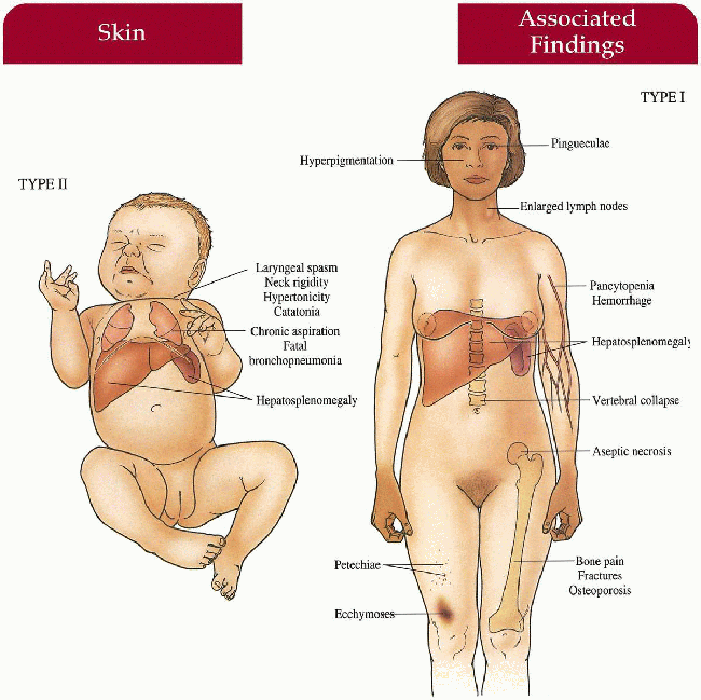
Type I (Adult)
Skin
May have diffuse hyperpigmentation on face, neck, hands; petechiae, ecchymoses
Musculoskeletal
Bone pain, fractures with thinned cortex, vertebral collapse, aseptic necrosis of femoral head
Gastrointestinal
Hepatomegaly, hypersplenism with secondary pancytopenia, hemorrhage
Lymphatics
Enlarged lymph nodes
Eyes
Pingueculae
Type II (Infantile) Central Nervous System
Hypertonicity, neck rigidity, laryngeal spasm, difficulty swallowing, catatonia, developmental retardation
Gastrointestinal
Hepatosplenomegaly
Lungs
Chronic aspiration, fatal bronchopneumonia
Type III
Rarest, rapidly deteriorates like type II; hepatosplenomegaly, bone involvement, strabismus, slow neurodegeneration
Laboratory Data
Serum glucocerebrosidase enzyme assay
Bone marrow biopsy—Gaucher cells
Bone x-rays
Serum acid phosphatase elevated
Management
Type II—supportive care, antibiotics
Type I—enzyme replacement, bone marrow transplant, splenectomy, referral to orthopedic surgeons for conservative management, referral to hematologist-oncologist
Prognosis
Type II—fatal by 1 to 2 years of age secondary to aspiration pneumonia
Type I—variable life span, potential premature death secondary to infection, anemia, hemorrhage without treatment; many may achieve a normal life span
Clinical Pearls
Infantile and adult forms may share mutation… Type II’s are kids with big spleens and livers who neurologically deteriorate rapidly… There’s essentially no residual enzyme in the infants… Type I’s always have some residual enzyme… The brain probably doesn’t need as much enzyme to dispose of the lipid… We are screening Ashkenazi Jews before pregnancy for both enzyme and DNA… Now we can treat symptomatic patients with enzyme… Livers and spleens shrink right up with the enzyme… There’s a joke that we have to provide the kids with suspenders along with their Ceredase because their pants start falling down… The enzyme can’t help the neuropathic forms (types II and III) because it doesn’t cross the blood-brain barrier… Bony problems improve with enzyme if one intervenes early on… All kids who present with hepatosplenomegaly are initially worked up with an enzyme assay. KH, JW
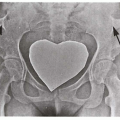
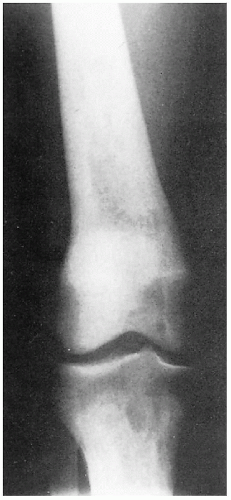 11.5. X-ray depicting tapered femoral midshaft with widening of the distal femur-characteristic “Ehrlenmeyer flask” deformity. (123) |
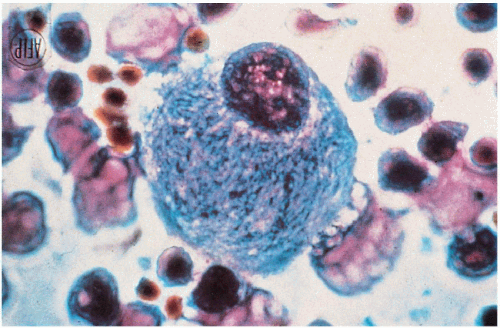 11.6. Gaucher cell—lipid-engorged macrophage with characteristic “crumpled tissue paper” appearance obtained from bone marrow. (48) |
|
Niemann-Pick Disease
Inheritance
Autosomal recessive; sphingomyelin phosphodiesterase-1 (SMPD-1) gene locus 11p15.4-15.1 (types A, B)
Type C—gene locus 18p
Prenatal Diagnosis
CVS/amniocentesis—sphingomyelinase enzyme assay from cultured chorionic villus tissue/amniotic fluid cells
DNA analysis
Incidence
Type A most common—over 50% are Ashkenazi Jews; a few hundred cases reported; M=F
Age at Presentation
Type A—infancy
Type B—infancy to childhood
Type C—childhood
Pathogenesis
Mutations in SMPD-1 results in acid sphingomyelinase deficiency in types A and B with subsequent accumulation of sphingomyelin in characteristic foam cells within all organs, increased in brain (except type B), liver, spleen, lymph nodes, and lungs; cholesterol esterification defect in type C with normal sphingomyelinase
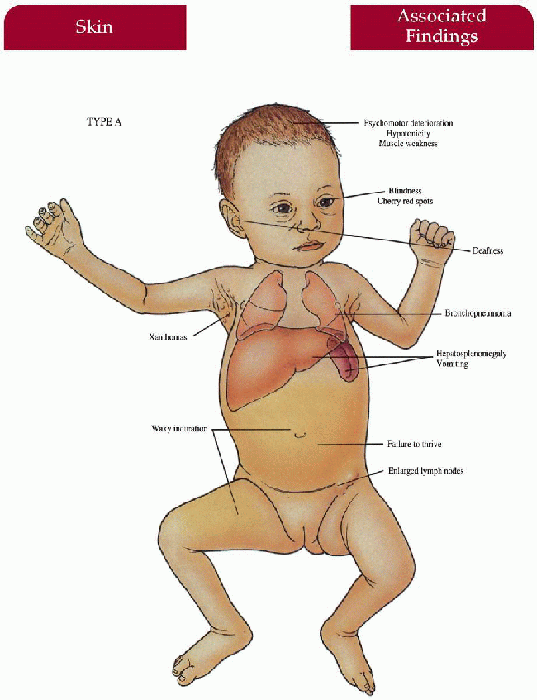
Key Features
Type A
Skin
Xanthomas; yellow-brown, waxy induration on exposed surfaces
Central Nervous System
Progressive psychomotor deterioration, hypotonicity, muscle weakness
Gastrointestinal
Hepatosplenomegaly, emaciated appearance—failure to thrive, vomiting
Lymphatics
Generalized enlarged lymph nodes
Eyes
Blindness, cherry red spots
Ear-Nose-Throat
Deafness
Lungs
Bronchopneumonia, infiltration of foam cells
Type B
CNS spared, otherwise similar to type A
Type C
Developmental delay, hepatosplenomegaly, progressive psychomotor deterioration
Laboratory Data
Serum sphingomyelinase assay
Bone marrow biopsy
Management
Supportive care—parenteral nutrition, antibiotics, transfusions, splenectomy
Bone marrow transplant—type B
Prognosis
Type A—death by 2 to 3 years of age secondary to progressive deterioration, fatal pulmonary infection
Type B—death in adolescence; may survive into adulthood
Type C—death in adolescence
Clinical Pearls
The gene has been cloned and most mutations in Ashkenazi Jews are pretty well known… We screen Ashkenazi Jewish couples preconceptually for carrier status… Same gene for types A and B, different mutations… Type B’s have higher residual enzyme and thus don’t get neurologic sequelae… B’s can get terrible lung disease… We have an ongoing study looking at replacement therapy in type B… Type C is a different gene seen in French-Canadians… The defect in C secondarily affects sphingomyelin… As in all babies with vomiting, their formulae are changed five times prior to diagnosis… Cherry red spots are also seen in Tay-Sachs, generalized sialidosis, and Sandhoff’s disease. KH, JW
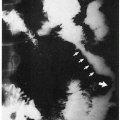
 11.7. Cherry red spot in fovea. (124) |
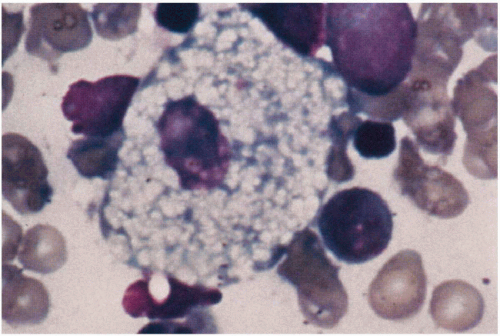 11.8. Niemann-Pick cell—foamy histiocyte obtained on bone marrow biopsy. (48) |
|
Mucopolysaccharidoses
Inheritance
Autosomal recessive except X-linked recessive in Hunter’s syndrome
Gene loci
Hurler, Scheie syndrome—4p16.3
Hunter’s syndrome—Xq27.3-q28
Sanfilippo syndrome—several loci reported
Morquio syndrome (A)—16q24.3
Maroteaux-Lamy syndrome—5q11-q13
Prenatal Diagnosis
CVS/amniocentesis—enzyme assay from cultured chorionic villus tissue/amniotic fluid cells
DNA analysis
Incidence
Hurler’s—approximately 1:100,000; M=F
Scheie’s—rare; M=F
Hunter’s—approximately 1:100,000; all males
Sanfilippo’s—approximately 1:25,000; M=F
Morquio’s—<1:100,000; M=F
Maroteaux-Lamy’s—rare; M=F
Age at Presentation
Hurler’s, Hunter’s, Sanfilippo’s, Morquio’s, Maroteaux-Lamy—normal at birth, within first 2 years of life Scheie’s—birth (corneal clouding, herniae); childhood (stiff joints)
Pathogenesis
Lysosomal enzymes responsible for breakdown of mucopolysaccharides are deficient; increased mucopolysaccharides throughout system
Syndrome: Enzyme; Mucopolysaccharides
Hurler’s, Scheie: α-L-iduronidase; dermatan, heparan sulfate
Hunter’s: iduronate sulfatase; dermatan, heparan sulfate
Sanfilippo’s: (A) heparan-N-sulfatase, (B) α-N acetylglucosaminidase, (C) acetyl-CoA: α-glucosaminide acetyltransferase, (D) N-acetylglucosamine 6-sulfatase; heparan sulfate
Morquio’s: (A) hexosamine 6-sulfatase, (B) beta-galactosidase; keratan sulfate
Maroteaux-Lamy’s: arylsulfatase B; dermatan sulfate
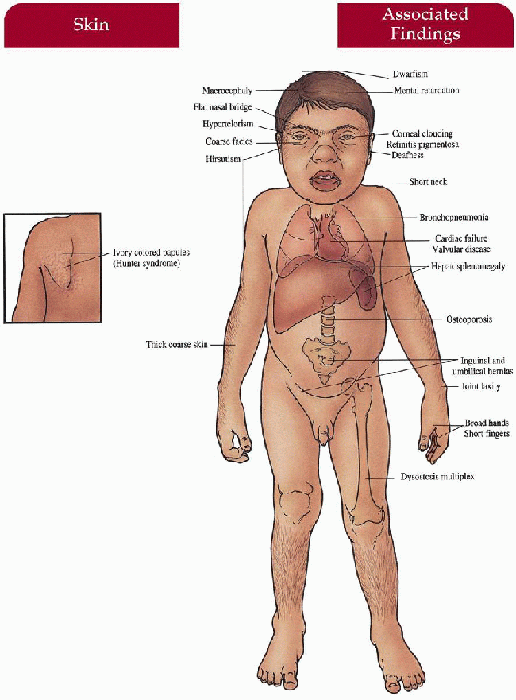
Key Features
Skin
Firm, ivory-colored papules symmetrically distributed between angles of the scapulae and posterior axillary line (Hunter)
Thick, coarse (all)
Hair
Generalized hirsutism (all)
Craniofacial
Coarse facies with thick nose and depressed nasal bridge, thick lips and tongue, short neck, macrocephaly (Hurler’s, Hunter’s, Sanfilippo’s—mild, Maroteaux-Lamy’s)
Central Nervous System
Mental retardation (Hurler’s, Hunter’s—severe form, Sanfilippo’s), progressive neurologic impairment (Hunter’s—severe form, Sanfilippo’s), deafness (Hurler’s, Hunter’s), hyperactivity/behavioral problems (Sanfilippo’s), hydrocephalus (Hurler’s, Hunter’s—severe form)
Musculoskeletal
Short stature (all except Scheie’s), broad hands with short fingers (all), “dysostosis multiplex”—stiff joints, contractures, kyphoscoliosis, claw deformity of hand (Hurler’s, Scheie’s, Hunter’s, Maroteaux-Lamy’s), odontoid hypoplasia (Morquio’s, Hurler’s), joint laxity (Morquio’s), lumbar lordosis (Morquio’s), umbilical/inguinal hernia (Hurler’s, Scheie’s, Hunter’s, Maroteaux-Lamy’s)
Eyes
Corneal clouding (all except Hunter’s, Sanfilippo’s)
Cardiovascular
Deposition of mucopolysaccharides with valvular and coronary heart disease (Hurler’s, Scheie’s, Hunter’s, Morquio’s, Maroteaux-Lamy’s), aortic valve disease (Scheie’s)
Gastrointestinal
Hepatosplenomegaly (Hurler’s, Hunter’s, Maroteaux-Lamy’s)
Lungs
Bronchopneumonia—often end-stage, sleep apnea with narrow upper airway (Hurler’s, Scheie’s, Hunter’s, Maroteaux-Lamy’s)
Differential Diagnosis
Mucolipidoses
Laboratory Data
Mucopolysaccharide detection in urine
Enzyme assay: fibroblasts, leukocytes, serum
Spine films
Echocardiogram
Management
Supportive care with physical therapy, special education, hearing aids
Surgical correction of cornea, cardiac valve, cervical spine, joint contractures, hernia may help
Bone marrow transplantation—some success
Prognosis
Progressive worsening with no cure; death usually within second decade because of respiratory/cardiac decompensation; milder forms may survive into adulthood Scheie’s—normal life span
Clinical Pearls
The coarse facies may not be apparent when they’re very young… The early tip off to the pediatrician is hepatosplenomegaly and a gibbous deformity of the lower spine… Early on, get a lateral lumbar spine and/or lateral skull seeing the j-shaped deformity of the sella turcica… Kids with Sanfilippo’s are physically very mild, but mentally can be difficult with hyperactivity, aggressive behavior… Four or five different enzyme defects can lead to Sanfilippo’s… As opposed to Hurler’s where different mutations in the same gene gives different disease… Most kids with storage disease seem to begin life reaching their early milestones and then drop off… If the mother tells you the kid isn’t doing what he’s supposed to be doing at 6 months or so, you really need to start looking… To obtain skin for fibroblast cultures, I (KH) take a tuberculin syringe, raise a bleb in the skin with lidocaine, and then come out the other end of the bleb… I lift it and shear off the base with a scalpel… Fibroblasts grow beautifully… We do echoes and spine films on all of them… As with Down’s, patients with Hurler’s and Morquio’s lack their occipito-atlantic prominence that anchors the cervical spine to the skull… They can split and become quadriplegic… You must fuse this area… We keep them in physical therapy to prevent contractures… The group in Minnesota has studied bone marrow transplants in all patients with mucopolysaccharidoses (MPS)… They believe it may mitigate the retardation… They’ve transplanted a kid with Hurler’s in utero… They may be slowing things down some but those cells are not going to correct the brain… You’re going to have to wait for gene therapy… Corneal transplants stay clear because they produce their own enzyme… The MPS society is very active… Gene replacement is being done on animal models like the Siamese cat for Maroteaux-Lamy’s. KH, JW
 11.9. Typical coarse facies in a girl with Hurler syndrome. |
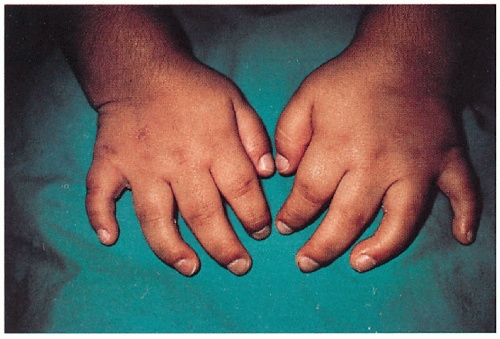 11.10. Broad hands with short fingers, thick coarse skin. (1) |
 11.11. Ivory-colored papules in “cobblestone” pattern between scapula and posterior axillary line in patient with Hunter’s syndrome. (125) |
|
Multiple Carboxylase Deficiency
Synonym
Biotinidase deficiency
Holocarboxylase synthetase deficiency
Inheritance
Both autosomal recessive; holocarboxylase synthetase (HLCS) gene on 21q22; biotinidase (BTD) gene on 3p25
Prenatal Diagnosis
CVS/amniocentesis: biotinidase or holocarboxylase synthetase assay
Incidence
Biotinidase deficiency: 1:70,000 to 80,000; M=F
Holocarboxylase synthetase deficiency: unknown, rare: M=F
Age at Presentation
Biotinidase deficiency: approximately 6 months old
Holocarboxylase synthetase deficiency: first few days to months of life
Pathogenesis
Mutations in HLCS or BTD render the patient deficient in holocarboxylase synthetase or biotinidase respectively, resulting in decreased free serum biotin and metabolic acidosis with resultant phenotype
Key Features
Skin
Periorificial/generalized dermatitis with/without candida infection
Hair
Sparse to total alopecia
Central Nervous System
Hypotonia, seizures, ataxia, coma
Gastrointestinal
Vomiting (holocarboxylase synthetase deficiency)
Eyes
Optic atrophy (biotinidase deficiency)
Ear-Nose-Throat
High-frequency hearing loss (biotinidase deficiency)
Metabolism
Metabolic acidosis, hyperammonemia, organic aciduria
Differential Diagnosis
Atopic dermatitis
Seborrheic dermatitis
Acrodermatitis enteropathica (p. 328)
Mucocutaneous candidiasis
Essential fatty acid deficiency
Laboratory Data
Screen urine for organic aciduria
Serum biotinidase/holocarboxylase synthetase assay
Screen blood—metabolic acidosis, hyperammonemia
Management
Biotin 10 mg per day for life
Prognosis
If biotin instituted prior to neurologic sequelae, normal life span with normal growth and development
Clinical Pearls
Clearly, we look for it in a kid who is acidotic in a precomatose state… We also look for all the other inborn errors that can do this… Any child who presents with failure to thrive and skin manifestations should to have their urine screened for organic acids. KH, JW
|
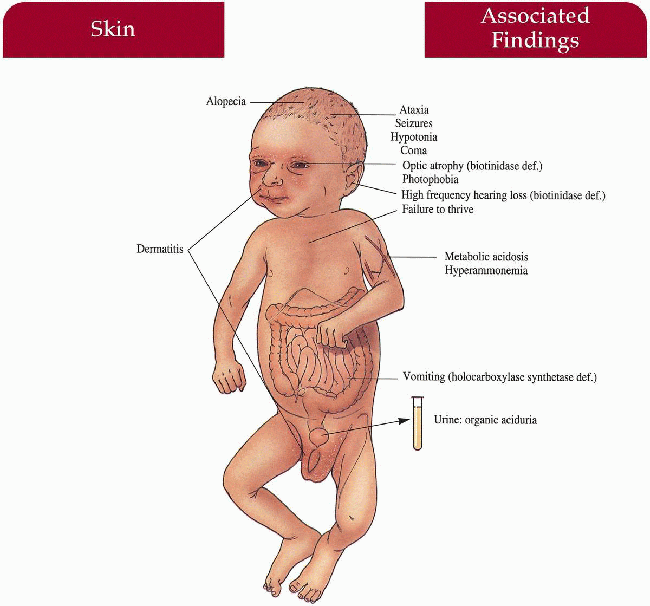
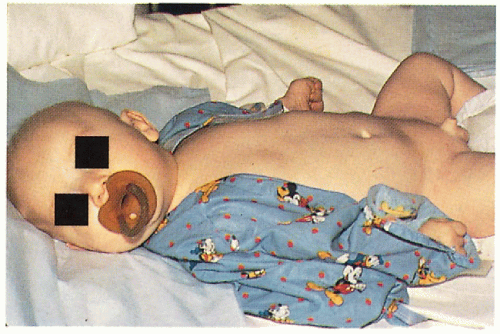 11.12. Infant with alopecia, hypotonia, and groin dermatitis. (5) |
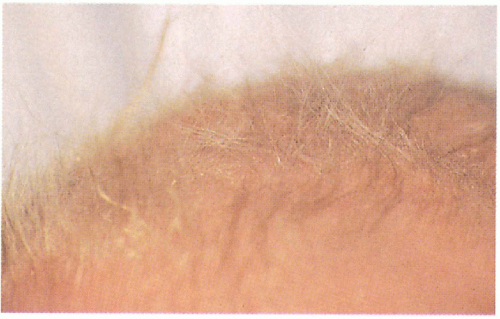 11.13. Sparse, lusterless, brittle scalp hair on close-up view. (5) |
Phenylketonuria
Inheritance
Autosomal recessive; phenylalanine hydroxylase (PAH) gene locus 12q24.1
Prenatal Diagnosis
CVS/amniocentesis—enzyme assay, metabolite levels, DNA analysis
Incidence
1:10,000 caucasian births; M=F
Age at Presentation
Birth
Pathogenesis
Mutation in PAH leads to a deficiency of phenylalanine hydroxylase with subsequent accumulation of phenylalanine and its metabolites; increased phenylalanine competitively inhibits tyrosine in melanogenesis and has toxic affects on the CNS
Key Features
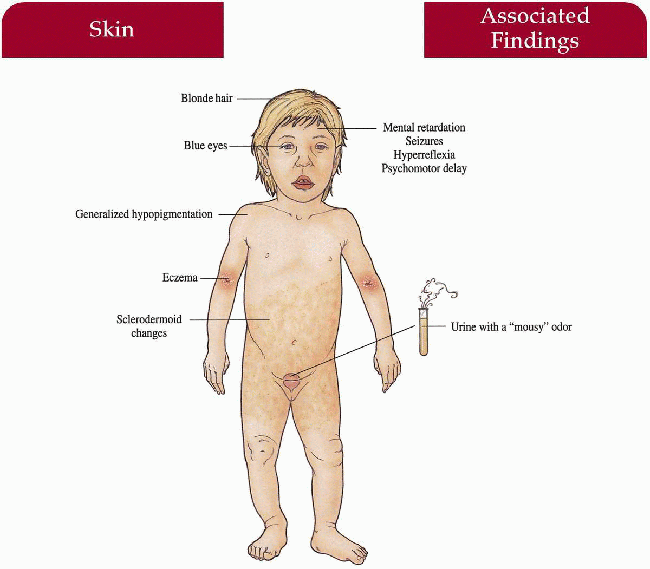
Skin
Generalized hypopigmentation, eczematous dermatitis, sclerodermoid changes
Hair
Blonde
Eyes
Blue
Central Nervous System
Mental retardation, seizures, hyperreflexia, psychomotor delay
Laboratory Data
Urine (mousy odor), blood screen for phenylalanine and its metabolites
Management
Routine neonatal screening
Low-phenylalanine diet instituted early on will prevent CNS and skin changes
Prognosis
Normal life span with normal intelligence with treatment; without treatment shortened life span with chronic seizures and mental retardation
Clinical Pearls
We don’t see it anymore… We’re screening them all at birth… I (KH) remember seeing a kid with phenylketonuria (PKU) who came from blonde-haired parents… He looked like an albino… Pregnant women with PKU must be on strict diet control to protect heterozygote fetus from malformations and severe retardation… Patients need to be on formula diet for rest of their lives… Tastes terrible… If the patient’s not under control, the mousy odor will be present… The enzyme is not expressed in cultured amniocytes… Prenatal diagnosis by molecular techniques if the mutation is known… People are using polymerase chain reaction (PCR) in situations where fibroblast or lymphocyte cultures don’t express enough enzyme to make the diagnosis. KH, JW
|
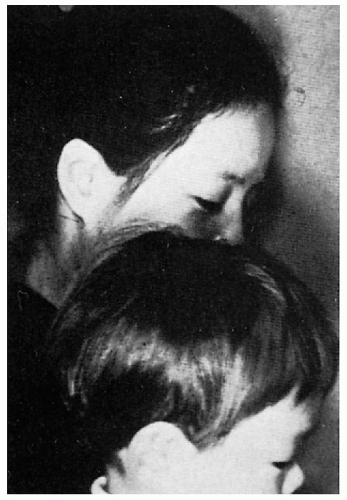 11.14. Lighter hair color in affected Japanese child compared to unaffected mother. (126) |
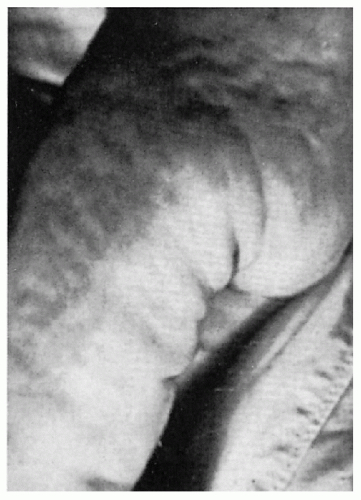 11.15. Sclerodermoid changes on buttocks and thigh of 1-year-old. (127) |
Wilson’s Disease
Synonym
Hepatolenticular degeneration
Inheritance
Autosomal recessive; ATB7B gene on 13q14.2-q21
Prenatal Diagnosis
DNA analysis
Incidence
1:50 to 100,000: M=F
Age at Presentation
Childhood to adulthood
Pathogenesis
Mutation in ATB7B, a gene encoding an adenosine triphosphatase (ATPase) CU2+-transporting polypeptide results in a defect in biliary excretion of copper/copper transport and leads to accumulation of copper in liver, brain, cornea
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


