Introduction332
INTRODUCTION
Normal elastic tissue
Structure and composition
Formation of elastic fibers
Degrading of elastic tissue
Age-related changes
Staining of elastic tissue
Categorization of elastic tissue disorders
Diagnosis
Clinical features
Pathology
Elastoma
Solitary or multiple; papules and disks; sometimes osteopoikilosis; linear variant reported
Increased, thick, branching elastic fibers
Linear focal elastosis
Palpable stria-like yellow lines; lumbosacral region
Numerous elongated wavy fibers, some with ‘paintbrush’ ends
Focal dermal elastosis
Late onset, PXE-like lesions
Increase in normal elastic fibers; no PXE changes
Elastoderma
Localized lax, wrinkled skin
Increased, pleomorphic elastic tissue in upper dermis
Elastofibroma
Deep scapular region; older age
Proliferation of collagen and elastic tissue
Elastosis perforans serpiginosa
Hyperkeratotic papules on face and neck
Papillary accumulation and transepidermal elimination of elastic tissue
Pseudoxanthoma elasticum (PXE)
Yellowish papules and plaques; angioid streaks
Fragmented and calcified elastic fibers in mid dermis; may perforate
Elastic globes
Asymptomatic
Basophilic cytoid bodies in the upper dermis
Solar elastosis
Thickened, furrowed skin
Accumulation of curled basophilic elastic fibers and elastic masses in upper dermis
Nodular elastosis
Usually periorbital with cysts and comedones
Comedones and usually solar elastosis
Elastotic nodules of the ears
Asymptomatic papules on the ear
Clumped masses of elastotic material
Collagenous and elastotic plaques
Waxy, linear plaques at juncture of palmar and dorsal skin
Thick collagen, some perpendicular; admixed granular, elastotic material; basophilic elastotic masses
Erythema ab igne
Follows repeated heat exposure
Elastotic material in dermis
Nevus anelasticus (papular elastorrhexis)
Papular lesions on lower trunk; early onset, no inflammation
A ‘minus nevus’ with reduced, fragmented elastic tissue in reticular dermis
Perifollicular elastolysis
Common; face and back; often associated acne vulgaris
Loss of elastic tissue around follicles
Anetoderma
Well-circumscribed areas of soft, wrinkled skin; may have preceding inflammation or be secondary to some other disease
Loss of elastic fibers, particularly in mid dermis
Papillary-dermal elastolysis (fibroelastolytic papulosis)
Papules and cobblestone plaques on neck and upper trunk, resembling PXE
Loss of elastic tissue in papillary dermis; no calcification of remaining fibers
Mid-dermal elastolysis
Widespread patches of fine wrinkling; additional perifollicular papules in some cases; may have preceding inflammatory phase
Loss of elastic tissue from mid dermis
Cutis laxa
Widespread, large folds of pendulous skin; often involves internal organs; congenital or acquired
Fragmentation and loss of elastic fibers
Menkes’ syndrome
Copper storage disease; brittle, ‘steel wool’ hair; vascular and neurological changes
Pili torti, often with monilethrix and trichorrhexis nodosa
‘Granulomatous slack skin’
Pendulous skin in flexural areas; T-cell lymphoma
Lymphoid cells; granulomas with multinucleate giant cells; absence of elastic fibers
Disease
Genetic abnormality (mutations unless stated)
Buschke–Ollendorff syndrome (elastomas)
Loss of function mutations in LEMD3 (MAN1) on chromosome 12q14
Pseudoxanthoma elasticum
ABCC6 gene on chromosome 16p13.1
Cutis laxa (dominant)
ELN (elastin) gene on chromosome 7q11.2
Williams–Beuren syndrome
Deletion of ELN and contiguous genes on chromosome 7q11.2
Cutis laxa (recessive)
Cutis laxa (X-linked recessive)
Allelic to Menkes’ syndrome (see below)
Cutis laxa (acquired)
FBLN5 and ELN mutations may predispose to some cases
Costello syndrome
Unknown; functional deficiency of elastin-binding protein
HRAS gene on chromosome 11p15.5
Mid-dermal elastolysis
One family with Keutel syndrome had this change. Defect in MGP (matrix Gla protein) gene
Wrinkly skin syndrome
Deletions of 2q32 reported, but not subsequently accepted. Defect in N-protein glycosylation likely
Marfan’s syndrome
Fibrillin-1 (FBN1) gene of 15q21.1
Menkes’ syndrome
ATP7A gene on chromosome Xq12–q13
Fragile X syndrome
FMR1 gene on chromosome Xq27.3
INCREASED ELASTIC TISSUE
ELASTOMA (ELASTIC NEVUS)
Histopathology
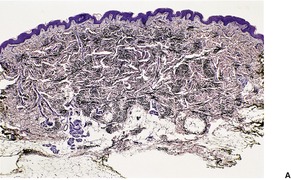

Fig. 12.1
Electron microscopy
LINEAR FOCAL ELASTOSIS
Histopathology
FOCAL DERMAL ELASTOSIS
Histopathology
ELASTODERMA
Histopathology
ELASTOFIBROMA
Histopathology
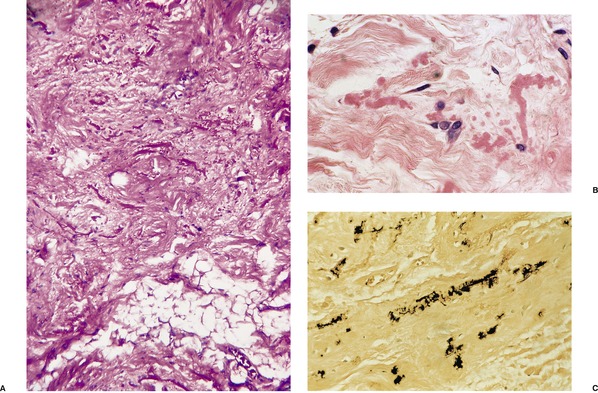
Fig. 12.2
ELASTOSIS PERFORANS SERPIGINOSA
Histopathology100.101.102. and 103.
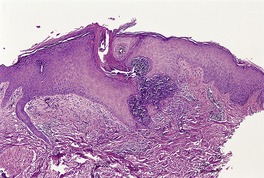
Fig. 12.3
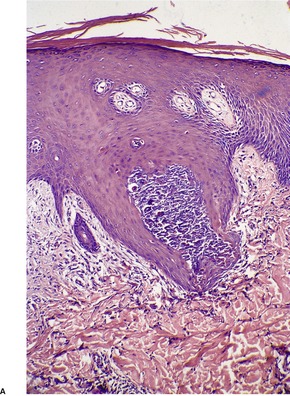
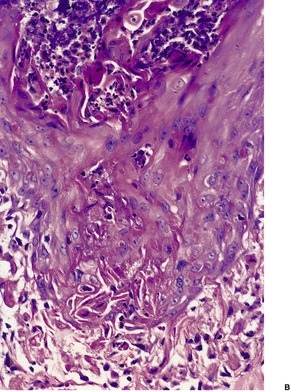
Fig. 12.4
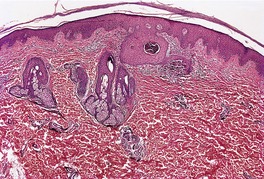
Fig. 12.5
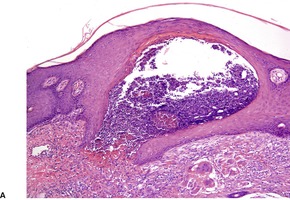
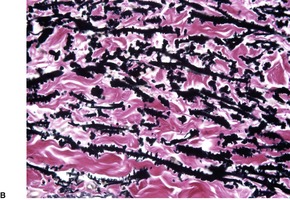
Fig. 12.6
Electron microscopy
PSEUDOXANTHOMA ELASTICUM
Acquired pseudoxanthoma elasticum
Histopathology147
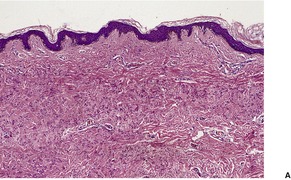
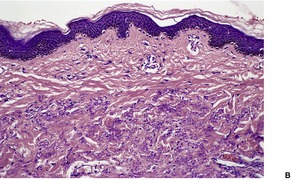
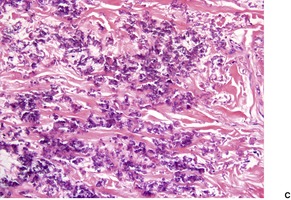
Fig. 12.7
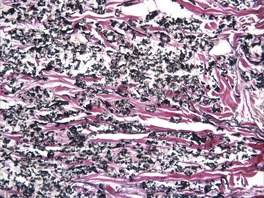
Fig. 12.8
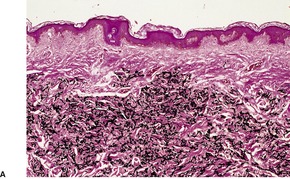
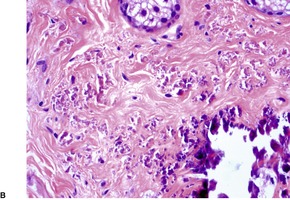
Fig. 12.9
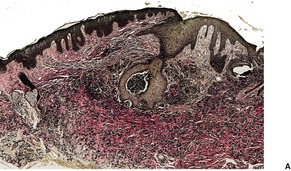
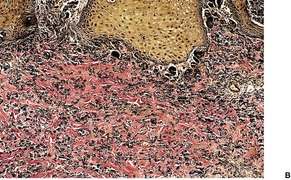
Fig. 12.10
Electron microscopy
ELASTIC GLOBES
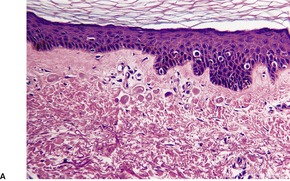
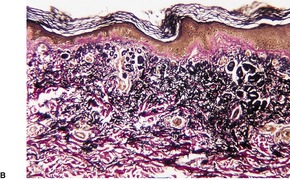
Fig. 12.11
SOLAR ELASTOTIC SYNDROMES
SOLAR ELASTOSIS (ACTINIC ELASTOSIS)
Histopathology
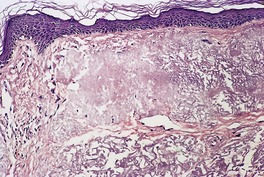
Fig. 12.12
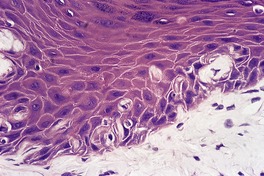
Fig. 12.13
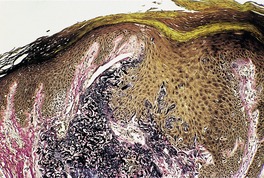
Fig. 12.14
Electron microscopy
NODULAR ELASTOSIS WITH CYSTS AND COMEDONES
Histopathology310. and 311.
ELASTOTIC NODULES OF THE EARS
Histopathology321
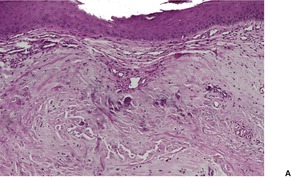
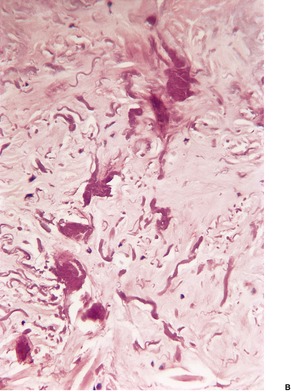
Fig. 12.15
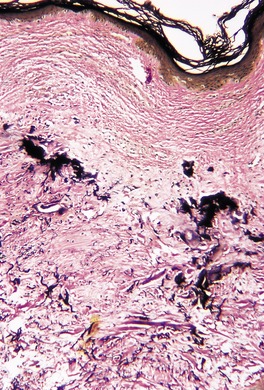
Fig. 12.16
COLLAGENOUS AND ELASTOTIC PLAQUES OF THE HANDS
Histopathology
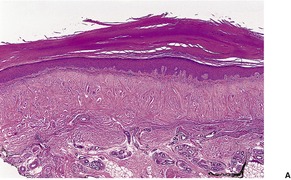
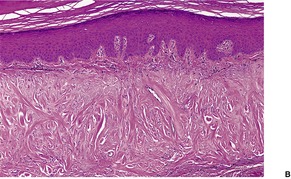
Fig. 12.17
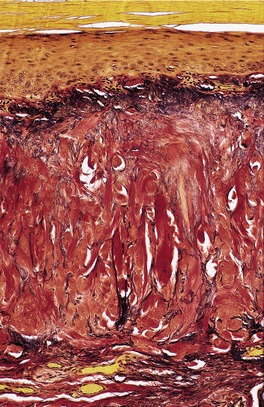
Fig. 12.18
ERYTHEMA AB IGNE
Histopathology330. and 337.
DECREASED ELASTIC TISSUE
NEVUS ANELASTICUS (PAPULAR ELASTORRHEXIS)
Histopathology
PERIFOLLICULAR ELASTOLYSIS
Histopathology348
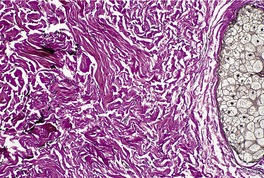
Fig. 12.19
ANETODERMA
Histopathology388
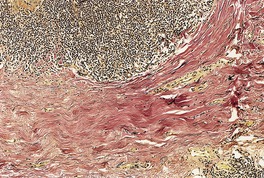
Fig. 12.20
CUTIS LAXA
Histopathology467
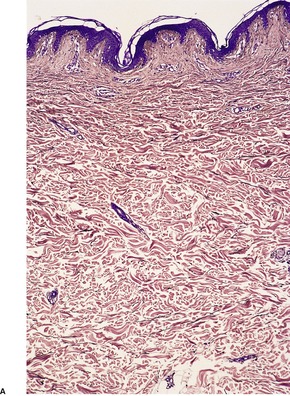
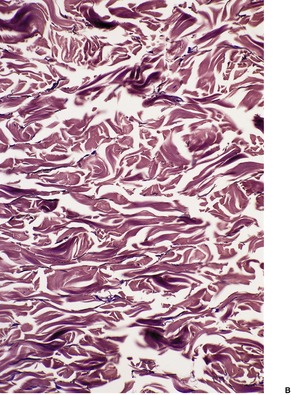
Fig. 12.21
Electron microscopy399
Elastolysis of the earlobes
WILLIAMS–BEUREN SYNDROME
Histopathology
PAPILLARY-DERMAL ELASTOLYSIS (FIBROELASTOLYTIC PAPULOSIS)
Histopathology
MID-DERMAL ELASTOLYSIS
Histopathology
MENKES’ SYNDROME
Histopathology
Electron microscopy
FRAGILE X SYNDROME
Histopathology522
WRINKLY SKIN SYNDROME
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Disorders of elastic tissue
Decreased elastic tissue344
Nevus anelasticus (papular elastorrhexis)344
Perifollicular elastolysis345
Anetoderma345
Williams–Beuren syndrome348
Papillary-dermal elastolysis (fibroelastolytic papulosis)348
Mid-dermal elastolysis348
Menkes’ syndrome349
Fragile X syndrome349
Wrinkly skin syndrome349
Granulomatous diseases349
‘Granulomatous slack skin’349
Myxedema350
Acrokeratoelastoidosis350
Elastic fibers are the important resilient component of mammalian connective tissue, and their presence is necessary for the proper structure and function of the cardiovascular, pulmonary, and intestinal systems.1. and 2. Their structural role is to endow tissues with elastic recoil and resilience. 3 They constitute less than 4% of the dry weight of the skin, forming a complex and extensive network in the dermis which imparts elasticity to the skin. 4
Mature elastic fibers are composed of structural glycoproteins, which contribute to the formation of 10–12 nm microfibrils, and elastin, a fibrous protein with a molecular weight of 72 000 kDa. 4 Elastin forms an amorphous core to the elastic fibers and this is surrounded by the microfibrils. Approximately 90% of the mature elastic fiber is elastin. It has a high concentration of alanine and valine, but less hydroxyproline than is present in collagen. Elastin-producing cells secrete tropoelastin, a 70 kDa precursor of elastin, which becomes highly crosslinked by the action of lysyl oxidase to form mature elastin. 5 It is the product of a single copy gene, located in the chromosomal locus 7q11.2, in the human genome. 6 Mutations in this gene cause the Williams–Beuren syndrome and some cases of cutis laxa. The microfibrils consist of several distinct proteins including fibrillin; abnormalities of the microfibrils occur in Marfan’s syndrome (see p. 350). The fibrillin-1 gene (FBN1), identified in 1991, is located on chromosome 15q21.1. 5 There is a second fibrillin gene on chromosome 5 (FBN2), mutations of which cause congenital contractural arachnodactyly. 7
A new family of extracellular matrix proteins, the fibulins, is being characterized progressively. Fibulins 1, 2, 3, 4, and 5 have been identified. The latter, fibulin-5, appears to be deficient in the autosomal recessive form of cutis laxa.8. and 9. Fibulin-5 is essential for dermal elastic fiber assembly. 10 The various fibulins are localized to different parts of the elastic fiber with fibulin-1 in the amorphous core and fibulin-2 in the fibrillin-containing elastin-associated microfibrils. Fibulin-2 is increased in solar elastosis. 11 Fibulin-4 is also essential for elastic-fiber formation, since its absence abolishes normal elastogenesis and leads to irregular elastin aggregates. 3 Its absence has also been found in a rare form of autosomal recessive cutis laxa. 12
The papillary dermis contains fine fibers which run perpendicular to the dermoepidermal junction and connect the basal lamina to the underlying dermal elastic tissue. 13 These oxytalan fibers, as they are called, consist of microfibrils without a core of elastin. 14 They branch to form a horizontal plexus in the upper reticular dermis, where they are known as elaunin fibers. They contain a small amount of elastin. The mature elastic fibers with their full composition of elastin are found below this in the reticular dermis. These three types of fibers probably correspond to consecutive stages of normal elastogenesis. 14
The formation of elastic fibers by fibroblasts, and in some circumstances by smooth muscle cells and chondroblasts, entails several different steps which are still poorly understood. Theoretically, these stages would include the expression of genes coding for elastin polypeptides, various intracellular processes, secretion of the precursor components, and extracellular modifications leading to the assembly of the fibers. 4 Fibulin-5 plays an important role in this assembly by acting as an adaptor molecule between elastin and the matrix scaffold. 15
Elastin is secreted in the form of a precursor, tropoelastin. This is ultimately crosslinked with desmosine to form stable elastin. 16 The formation of desmosine requires the copper-dependent enzyme lysyl oxidase. 16 Defects in this enzyme can result from a spectrum of mutations in the ATPase gene (ATP7A), as seen in Menkes’ syndrome (see p. 349). Impaired elastinogenesis can also result from other altered transport mechanisms important to elastic fiber assembly. 7 One such example is a deficiency in elastin binding protein (EBP), which transports tropoelastin from its site of synthesis in the cell to the cell membrane. Costello syndrome (see p. 347) results from a functional deficiency in EBP.
A congenital disorder of glycosylation involving a defect in the biosynthesis of N- and O-glycans has been reported in several patients with cutis laxa, indicating the role these glycans play in the stability of various extracellular matrix proteins; they may be involved in the glycosylation of fibulin-5. 17
Very few enzymes can degrade crosslinked elastin. 18 One of these is elastase, which is found in the pancreas and in neutrophils, macrophages, platelets, certain bacteria, and cultured human fibroblasts.16. and 18. Elastases exhibit a broad specificity. They are found in all classes of proteinases. Elastase activity is present in neutrophil elastase, cathepsin G, proteinase 3, and matrix metalloproteinases 2 and 9 (gelatinases A and B). 19 MMP12 (metalloelastase) is another matrix metalloproteinase which also has an important role in the degradation of elastic fibers. 3 The exact role of elastase in normal skin is uncertain; it plays a part in the elastolysis seen in anetoderma and in acquired cutis laxa associated with inflammatory skin lesions. Elastase inhibitors also exist; these include α1-antitrypsin, α2-macroglobulin, and lysozyme. 20 There are two factors, vitronectin and delay-accelerating factor, which appear to prevent damage to elastic fibers by complement. 21 Further work is needed to clarify the role of these substances.
There is evidence of continuing synthesis of elastic fibers throughout life, but after the age of 50 the new fibers are loosely rather than closely assembled. 22 With age, there is some loss of the superficial dermal fibers and a slow, progressive degradation of mature fibers.16. and 23. This is accompanied by changes in collagen and extracellular matrix.24. and 25. Ultrastructural changes include the formation of cystic spaces and lacunae, imparting a porous look to the fibers;26. and 27. they may fragment or develop a fuzzy, indistinct border. 26 The changes are quite distinct from those seen in solar elastosis.
Another age-related change is the deposition on elastic fibers of terminal complement complexes and vitronectin. This latter substance is a multifunctional glycoprotein that is hypothetically involved in the prevention of tissue damage in proximity to local complement activation. 28
Elastic tissue can be demonstrated in hematoxylin and eosin-stained sections if appropriate modifications, as described by O’Brien, are made. 29 Notwithstanding this method, the commonly used stains for elastic tissue are the orcein, aldehyde–fuchsin, Verhoeff, and Weigert methods. However, the superficial fine elastic fibers do not stain with most of these methods, although they will with a modified orcein stain16 and the Luna stain, which incorporates aldehyde–fuchsin and Weigert’s iron hematoxylin. 30 The Luna stain also demonstrates a fibrillary component in solar elastosis. Elastic fibers stain a brilliant purple against a pale lavender background with this stain. 30 Miller’s modification of Weigert’s resorcin–fuchsin has been suggested as the best method for demonstrating new elastic fibers. 31
A monoclonal antibody, HB8, has been described as a stain for elastic fibers. 32 It has no advantages over the modified orcein, Luna, or Miller stains.
A simple classification of disorders of cutaneous elastic tissue divides them into those in which the elastic tissue is increased and those in which it is reduced. 33 The solar elastotic syndromes are best considered as a discrete group. Minor alterations in elastic tissue may occur in the various collagen disorders, in line with the observation that alterations in one component of the connective tissue matrix may influence the structure and function of others. 34 This group will not be considered in great detail here.
Although not categorized separately in this chapter, it should be remembered that elastic fibers are the most important structure to undergo transepidermal elimination. This can occur in elastosis perforans serpiginosa, perforating folliculitis, perforating pseudoxanthoma elasticum, solar elastosis, keratoacanthoma, healing wounds, and hypertrophic discoid lupus erythematosus.
The clinical and pathological features of the major disorders of elastic tissue are summarized in Table 12.1. The genetic abnormalities in the various disorders are listed in Table 12.2.
FBLN5 (fibulin-5) gene on chromosome 14
FBLN4 (fibulin-4) gene
Disorders of glycosylation
Very little is known about the mechanisms which lead to an increase in dermal elastic tissue. Besides the conditions to be considered below, a mild increase in elastic tissue has been reported in osteogenesis imperfecta, 35 chronic acidosis, 36 amyotrophic lateral sclerosis37 and some stages of radiation dermatitis.38. and 39. The solar elastotic syndromes are also characterized by increased elastic tissue and they will be considered after this section.
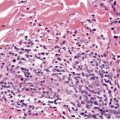
Elastoma (elastic nevus, juvenile elastoma, 40 nevus elasticus, connective tissue nevus of Lewandowsky type41) is a variant of connective tissue nevus (see p. 317) in which the predominant abnormality is an increase in dermal elastic tissue. 42 The lesions may be solitary or multiple40. and 43. and in the latter circumstance they are often associated with multiple small foci of sclerosis of bone (osteopoikilosis). This association is known as the Buschke–Ollendorff syndrome44. and 45. and the cutaneous lesions as dermatofibrosis lenticularis disseminata. In several instances, the cutaneous lesions have shown abnormalities in collagen rather than elastic tissue (collagenomas)46.47. and 48. and for this reason dermatofibrosis lenticularis disseminata is not entirely synonymous with the term ‘elastoma’. 43
The Buschke–Ollendorff syndrome (OMIM 166700) is inherited as an autosomal dominant trait with variable expressivity. 44 While most individuals with this syndrome have both skin manifestations and osteopoikilosis, some family members have only cutaneous lesions or only bony lesions, but not both.46.49.50.51. and 52. The genetic defect in the Buschke–Ollendorff syndrome has now been determined. It involves loss-of-function mutations in the LEMD3 gene (also called MAN1), which encodes an inner nuclear membrane protein that influences Smad signaling.53. and 54. Melorheostosis (OMIM 155950) appears to be allelic with this syndrome and the concurrence of the two diseases has been reported.54.55. and 56. The cutaneous lesions (elastomas) may be widely distributed flesh-colored or yellowish papules or localized, asymmetrically distributed plaques on the lower trunk or extremities. 57 These localized lesions are thought to represent type 2 segmental manifestations of an autosomal dominant trait. 58 A large, multilobulated, exophytic variant has been reported. 59 They develop at an early age. The nail–patella syndrome, another disorder of connective tissue, has been reported in a family with the Buschke–Ollendorff syndrome. 60
Studies of the desmosine content of elastomas indicate a 3–7-fold increase in elastin. 61 There appears to be an abnormality of elastogenesis with faulty aggregation of elastin units associated with the overall increase in elastin.
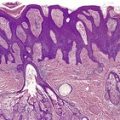
Examination of hematoxylin and eosin-stained sections usually shows a normal dermis, 44 although sometimes there is an increase in its thickness. The epidermis may have a slight wavy pattern. Elastic tissue stains show an accumulation of broad, branching and interlacing elastic fibers in the mid and lower dermis (Fig. 12.1). 47 The papillary dermis is unaffected. Sometimes the elastic fibers encase the collagen in a marble-vein configuration.40. and 62. Clumped elastic fibers have been reported; 63 they are a regular feature in linear focal elastosis. 64 A morphometric analysis shows a 4–5-fold increase in elastic fibers compared to normal skin; the diameter of the fibers is also increased. 65
Elastoma. (A) There is increased elastic tissue in much of the dermis, but excluding either end. (Orcein) (B) There are coarse irregular clumps of elastic tissue within the reticular dermis. (Verhoeff–van Gieson)
Uncommon changes include an increase in acid mucopolysaccharides, 47 slight thickening of collagen bundles, or a well-developed vascular component. 66 Two cases have been reported with facial plaques and increased dermal elastic tissue;67. and 68. in one, there was also perifollicular mucin. 68
Ultrastructural findings have been variable.61. and 69. Usually there are branched elastic fibers of variable diameter, without fragmentation. Elastic microfibrils may be replaced by granular or lucent material.40. and 70. Collagen fibers are sometimes increased in diameter69 and some fibroblasts may have dilated rough endoplasmic reticulum. 61 In linear focal elastosis, sequential maturation of elastic fibers can be seen, suggesting active elastogenesis. 71
Linear focal elastosis (elastotic striae) is a distinctive acquired lesion composed of palpable, stria-like, yellow lines that typically occur in the lumbosacral region,64.71.72.73.74.75.76. and 77. but other sites, such as the face78 and legs79 may be affected. There is a predilection for males. It occurs at all ages, but predominantly in the elderly. 80 This condition, which has been likened to a keloid of elastic fibers, may be an unusual form of striae distensae (see p. 319), 75 but its pathogenesis is currently unknown. 80
Focal dermal elastosis is a distinct entity of late onset, characterized by a pseudoxanthoma elasticum-like eruption.81.82.83. and 84. The elastin content of the skin is significantly increased. 85
There is an increase in normal-appearing elastic fibers in the mid and deep dermis. 81 There are no changes of pseudoxanthoma elasticum.
Elastoderma, an exceedingly rare condition, is an acquired, localized laxity of skin resembling cutis laxa with lax, extensible, wrinkled skin.86.87. and 88. The lesions are not indurated. The clinical presentation differs, therefore, from elastoma (elastic nevus).
Elastoderma has an excessive accumulation of pleomorphic elastic tissue within the dermis, particularly the upper dermis. On routine H & E sections, there are numerous eosinophilic irregular fibers in the papillary and reticular dermis. On elastic tissue stains, there are masses of thin, intertwined fibers. 86 The fibers are not calcified. 88
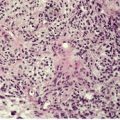
Elastofibroma (elastofibroma dorsi) is a relatively rare, slowly growing proliferation of collagen and abnormal elastic fibers with a predilection for the subscapular fascia of older individuals, particularly females.84.89.90.91. and 92. It is rarely found at other sites. Most elastofibromas are unilateral and asymptomatic. Multiple elastofibromas are rare. 93 Nearly two-thirds of the 300 or more cases so far reported have been from southern Japan. 94 Although an association with pseudoxanthoma elasticum has been recorded, there is no abnormality in the ABCC6 gene. 95 The pathogenesis is unknown, but they may represent a reaction to prolonged mechanical stress, possibly involving disturbed elastic fibrillogenesis by periosteal-derived cells. 96 Cytogenetic studies have found evidence of chromosomal instability and/or clonal changes suggesting a neoplastic process. 97 Elastofibromas are gray-white or tan in color and measure 5–10 cm in diameter. Subclinical elastofibromas have been found at autopsy. 98
Elastofibromas are non-encapsulated lesions which blend with the surrounding fat and connective tissue. 90 They are hypocellular and composed of swollen collagen bundles admixed with numerous, irregular, lightly eosinophilic fibers and some mature fat (Fig. 12.2). The fibers, which account for almost 50% of the tissue, stain black with the Verhoeff elastic stain. Some fibers are branched while others show a serrated edge.
Elastofibroma dorsi. (A) Coarse elastic fibers are admixed with collagen and adipose tissue. (B) The fibers have an irregular outline. (H & E) (C) An elastic tissue stain confirms the irregular outline. (Verhoeff–van Gieson)
The elastic fibers contain elastin but not fibrillin-1. No staining for actin or desmin was observed in one study. 95 The cells express CD34. 97
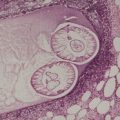
Elastosis perforans serpiginosa (also known as perforating elastosis – OMIM 130100) presents as small papules, either grouped or in a circinate or serpiginous arrangement, on the neck, upper extremities, upper trunk, or face.100.101.102.103.104.105. and 106. Rarely, the lesions are generalized.84.107. and 108. There is a predilection for males, with the onset usually in the second decade. Familial cases have been reported.109.110. and 111. An autosomal dominant mode of inheritance with variable expressivity of the trait has been suggested. 112 In up to a third of cases there is an associated systemic condition or connective tissue disorder: these include Down syndrome,108.113.114.115. and 116. osteogenesis imperfecta, 117 cutis laxa, 118 Ehlers–Danlos syndrome, Marfan’s syndrome, acrogeria, scleroderma,119. and 120. an abnormal 47,XYY karyotype, 121 diabetes mellitus, 122 perforating folliculitis, 123 and chronic renal failure. 124
Similar cutaneous lesions have been reported in patients with Wilson’s disease and cystinuria receiving long-term penicillamine therapy.118.125.126.127.128. and 129. In these patients, a local copper depletion or a direct effect of penicillamine on elastin synthesis may be responsible for the formation of the abnormal elastic fibers, which are then eliminated transepidermally.125. and 130. Elastic tissue damage appears to occur in other organs as well, a feature generally lacking in the usual idiopathic form of the disease.130. and 131. The nature of the defect in the idiopathic form is unknown, but it is possible that perforating elastosis is the final common pathway for more than one abnormality of elastic fibers.101. and 132. This theory is compatible with the finding of a 67 kDa elastin receptor in keratinocytes immediately surrounding the elastic materials being eliminated in lesions of elastosis perforans serpiginosa.133. and 134. The elastin receptor may be involved in the interaction between keratinocytes and elastin.133. and 134.
Various therapies have been used including liquid nitrogen cryotherapy, isotretinoin, and imiquimod. 135
In fully developed lesions there is a localized area of hyperplastic epidermis, associated with a channel through which the basophilic nuclear debris and brightly eosinophilic fragmented elastic fibers are being eliminated (Fig. 12.3). A keratinous plug usually overlies this channel, which may take the form of a dilated infundibular structure or a more oblique canal coursing through hyperplastic epidermis, follicular epithelium or the acrosyringium (Fig. 12.4 and Fig. 12.5). When the canal is oblique, sections may only show a surface plug of keratinous debris and a localized area of hyperplastic epidermis which in its lower portion forms a bulbous protrusion into the dermis. This appears to envelop an area of the papillary dermis containing basophilic debris and some refractile eosinophilic elastic fibers.
Elastosis perforans serpiginosa. Debris is entering a channel within the epidermis. (H & E)
(A) Elastosis perforans serpiginosa. (B) Debris and elastic fibers are being enveloped by a bulbous protrusion of the epidermis. (H & E)
Elastosis perforans serpiginosa. Another case with a less obvious epidermal channel. (H & E)
Elastic tissue stains show increased numbers of coarse elastic fibers in the papillary dermis. Some of these appear to overlap the basal epidermal cells. In the region of their transepidermal elimination, the elastic fibers lose their staining properties as they enter the canal and become brightly eosinophilic. They will stain with the Giemsa method. A few foreign body giant cells and inflammatory cells are often present in the dermis adjacent to the channel. In older lesions, there is focal dermal scarring and usually an absence of elastic fibers.
IgM, C3, and C4 were demonstrated on the abnormal elastic fibers in the papillary dermis in one of two cases studied by immunofluorescence. 136
In penicillamine-related cases, there is an increased number of thickened elastic fibers in the reticular dermis and less hyperplasia of elastic fibers in the papillary dermis, except in the areas of active transepidermal elimination. 137 The elastic fibers are irregular in outline with buds and serrations. This may be discerned in hematoxylin and eosin-stained preparations, but it is well shown by elastic tissue stains (Fig. 12.6) 138 or in Epon-embedded thin sections stained with toluidine blue. 130
(A) Elastosis perforans serpiginosa due to penicillamine therapy. (H & E) (B) The elastic fibers have a serrated appearance. (Verhoeff–van Gieson)
Ultrastructural examination of the dermis in penicillamine-related cases shows that the elastic fibers have a normal core and an irregular coat with thorn-like protrusions at regular intervals, the so-called ‘lumpy bumpy’ or ‘bramble-bush’ fibers.137.138.139. and 140. Collagen fibers are also abnormal with extreme variations in thickness.140. and 141. Electron microscopy of idiopathic cases has shown increased numbers of large elastic fibers which are convoluted and branching. 142 Fine filaments, similar to those in embryonic elastic fibers, are present on the surface of the fibers.142. and 143.
Pseudoxanthoma elasticum (OMIM 264800) is an inherited disorder of connective tissue in which calcification of elastic fibers occurs in certain areas of the skin, eyes, and cardiovascular system.144.145.146.147.148. and 149. Mutations in the ABCC6 gene on chromosome 16p13.1 are responsible for this condition. Over 60 different mutations of this gene have been reported.150. and 151. Its incidence ranges from 1 : 70 000 to 1 : 100 000 live births. 152 Skin changes are usually evident by the second decade and consist of closely set yellowish papules with a predilection for flexural creases, particularly in the neck and axillae and less commonly in the groins, periumbilical area, and the cubital and popliteal fossae.145.146.147.153. and 154. Oral lesions may occur. 148 The skin becomes wrinkled and thickened and eventually may become lax and redundant, resembling cutis laxa.155.156.157. and 158. Mental (chin) creases are common.159. and 160. The calcium content of affected skin may be up to several hundred times normal. 161 Calcification occurring in the breast may lead to problems in the interpretation of mammograms. 162 Eye changes include angioid streaks and a degenerative choroidoretinitis which may lead to blindness. Angioid streaks can occur in the absence of pseudoxanthoma elasticum. 163Calcification of elastic fibers in arteries and intimal and endocardial fibroelastosis develop. The vascular changes may lead to hypertension, sudden cardiac death, 164 cerebrovascular accidents, and gastrointestinal hemorrhage.165.166. and 167. Gastric bleeding may be increased in pregnancy, but most pregnancies in this condition are uncomplicated. 168
Pseudoxanthoma elasticum has been associated with certain hemoglobinopathies such as β-thalassemia and sickle cell disease.152. and 169. Nephrolithiasis is another rare association. 170 Its occurrence is interesting as the ABCC6 gene encodes a transmembrane transporter protein ABCC6 (MRP6), a member of the ATP-binding cassette (ABC) superfamily. 171 This protein is strongly expressed in the liver and kidney, with a possible role in phosphocalcic metabolism. 170 Hyperphosphatasia has been reported in several cases. 172 The administration of vitamin D3 results in further deposition of calcium salts. 173 Oral phosphate binders have been used in a small number of cases, with clinical improvement of skin lesions in half. 174 Urinary glycosaminoglycan levels are elevated early in the disease. 175 Polymyositis and lupus erythematosus are other associations, possibly due to chance.176. and 177.
Characterization of the gene mutations responsible for this condition have led to a change in our understanding of its inheritance. Previously it was believed that there were two variants with autosomal dominant inheritance and two with autosomal recessive inheritance. The clinical presentations and severity of these variants appeared to differ.178.179.180. and 181. It was acknowledged that the interpretation of the genetic studies that resulted in these views was made difficult by the limited phenotypic expression of the disease in some individuals. 182 It is now believed that fewer than 2% of cases, if any, are autosomal dominant. In one study of 142 subjects, all cases were autosomal recessive in their mode of inheritance. 183 Consanguinity was high. Homozygous patients had typical clinical appearances, including angioid streaks in the eye, although the severity of the disease varied. 183 All 67 subjects that were heterozygous for the gene showed no cutaneous features of pseudoxanthoma elasticum, but few had skin biopsies. 183 Heterozygotes may uncommonly experience severe ophthalmological complications. 123 Whether they may have cardiovascular complications remains to be determined. Another study showed histological changes in dermal elastic fibers in heterozygotes but the authors still concluded that the relevance of performing a skin biopsy to detect heterozygous carriers remains to be determined. 171
Patients purported to have coexisting elastosis perforans serpiginosa and pseudoxanthoma elasticum have been reported; they are now regarded as having perforating pseudoxanthoma elasticum.184.185.186. and 187. In such a case, associated with Moya Moya vasculopathy, amino acid substitutions were found in a region close to the ABCC6 gene site. 187 Many of these patients have so-called ‘acquired pseudoxanthoma elasticum’ (see below).
The factors that lead to the calcification of initially normal elastic fibers in pseudoxanthoma elasticum are not known. 188 The role of ABCC6 has not been delineated, and its substrate has not been identified. 171 Polyanionic material is deposited in association with the calcified material. Cultured fibroblasts from patients with this condition release a proteolytic substance and it has been postulated that this may cause selective damage to elastin, leading to calcification. 189 Fibrillin appears to be abnormal in only isolated cases (unlike the findings in Marfan’s syndrome). 190 Decreased deposition of fibrillin-2 has been reported in pseudoxanthoma elasticum. 191 These isolated findings have been replaced with a viewpoint that several structural proteins with an affinity for calcium (vitronectin, fibronectin, and bone sialoprotein) are increased in this condition and responsible for the calcification of the elastic fibers. 171
There has been one report of spontaneous resolution and repair of elastic tissue calcification. 192 There is one report of the successful treatment of this condition with oral tocopherol acetate and ascorbic acid, both antioxidants. 193 It took 2 years of treatment for the papules to disappear. 193
Acquired pseudoxanthoma elasticum refers to an etiologically and clinically diverse group of patients with late onset of the disease, no family history, absence of vascular and retinal stigmata, and identical dermal histology.149.194. and 195. The term ‘perforating calcific elastosis’ has been suggested for some of these cases.196. and 197. Included in this group are individuals exposed to calcium salts, including farmers exposed to Norwegian saltpeter (calcium and ammonium nitrate),198. and 199. and obese, usually multiparous black women who develop reticulated and atrophic plaques and some discrete papules around the umbilicus186.196. and 200. or lower chest. 201 Perforation is common in this latter group. 202 Patients with chronic renal failure on dialysis have also been reported with this acquired variant.194.203. and 204.
Pseudoxanthoma elasticum-like fibers have been found in other skin diseases in the absence of other signs of this disease,205. and 206. and as an acquired localized disorder. 207 These changes have been seen particularly in disorders of the subcutis such as lipodermatosclerosis, morphea profunda, erythema nodosum, but also in granuloma annulare, lichen sclerosus, tumefactive lipedema, and a basal cell carcinoma.205. and 206.
Pseudo-pseudoxanthoma elasticum refers to the development of the systemic changes of pseudoxanthoma elasticum in patients on long-term penicillamine therapy for Wilson’s disease. However, it has also been used for the cases referred to above as acquired pseudoxanthoma elasticum. 208 A spectrum of elastic tissue disorders, particularly elastosis perforans serpiginosa (see above), occurs with penicillamine therapy.209. and 210.
There are short, curled, frayed, basophilic elastic fibers in the reticular dermis, particularly in the upper and mid parts (Fig. 12.7 and Fig. 12.8). The papillary dermis is spared except at sites of transepidermal elimination (perforation). The elastic fibers in affected skin are stained black with the von Kossa method (Fig. 12.9). They stain with the Verhoeff method and there is intense blue staining with phosphotungstic acid hematoxylin (PTAH). Calcinosis cutis211 and osteoma cutis157. and 158. are rare complications. There is a good correlation between the severity of the clinical change and the histology. 183
(A) Pseudoxanthoma elasticum. (B) Note the short, curled elastic fibers in the reticular dermis. (C) They are basophilic. (H & E)
Pseudoxanthoma elasticum. The elastic fibers are short and curled. (Verhoeff–van Gieson)
Pseudoxanthoma elasticum. (A) Calcium salts are deposited on the abnormal elastic fibers. (von Kossa) (B) Larger deposits of dystrophic calcification may occur. (H & E)
If perforation is present, there is a focal central erosion or tunnel with surrounding pseudoepitheliomatous hyperplasia or prominent acanthosis (Fig. 12.10). Basophilic elastic fibers are extruded through this defect. Sometimes foreign body giant cells, histiocytes, and a few chronic inflammatory cells are present when there is perforation or traumatic ulceration.212. and 213. The giant cells may then engulf some elastic fibers.
(A) Perforating pseudoxanthoma elasticum. (B) The elastic fibers which are about to undergo transepithelial elimination are short, curled and frayed. Clumped elastic fibers are also present. (Verhoeff–van Gieson)
In the acquired localized forms the changes can be found in the dermis and/or the septa of the subcutaneous fat. 206
Calcification occurs initially in the central zones of the elastic fibers. 214 There is also some calcification of intercellular spaces and occasionally also of collagen fibers; the latter change may be reversible.172. and 201. There is continuing elastogenesis with some normal elastic fibers. 172 Twisted collagen fibrils and thready material, 215 which has been found to contain fibrinogen, collagenous protein, and glycoprotein, are also present. 216 This indicates that the abnormality is not limited to the elastic fibers.
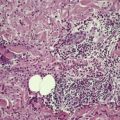
Elastic globes are small basophilic bodies, found in the upper dermis of clinically normal skin, which stain positively for elastic fibers (Fig. 12.11). They are considered with the other dermal cytoid bodies on page 387. Numerous elastic globes have been reported in a patient with epidermolysis bullosa whose skin was wrinkled217 and in a patient with the cartilage–hair hypoplasia syndrome whose skin was hyperextensible. 218
Elastic globes. (A) There are multiple, round, and ovoid deposits in the papillary dermis. Solar elastosis is also present. (H & E) (B) An elastic tissue stain of the same case. (Verhoeff–van Gieson)
As the globes contain D-aspartyl residue-containing peptide, it has been postulated that they result from UV-induced skin damage. 219
The term ‘solar elastosis’ refers to the accumulation of abnormal elastic tissue in the dermis in response to long-term sun exposure. Photoaging is a process distinct from the changes taking place due to chronological aging, although photoaging does increase in severity with chronological aging.220.221.222. and 223. The effects of both are cumulative. 224 A review of photoaging was published in 2007. 225 Another review looking at treatment options was published in 2008. 226 The cosmetic effects of photodamage are assuming increasing importance in society. There are many different clinical patterns of solar elastosis, some of which form distinct clinicopathological entities.227. and 228. Other clinical patterns are histologically indistinguishable from one another and they are usually grouped together under the umbrella term ‘solar elastosis’.
The following entities are regarded as solar elastotic syndromes:
• solar elastosis
• nodular elastosis with cysts and comedones
• elastotic nodules of the ears
• collagenous and elastotic plaques of the hands.
Colloid milium can also be regarded as a solar elastotic syndrome, as it appears that the colloid substance derives, at least in major part, from elastic fibers through actinic degeneration. 229 Colloid degeneration (paracolloid, colloid milium-like solar elastosis) has overlapping features histologically with both colloid milium and solar elastosis. These topics are considered with the cutaneous deposits in Chapter 14 (pp. 384 and 385).
Solar elastotic skin is more susceptible than normal skin to chronic infections with Staphylococcus aureus and several other bacteria. This results from a decline in the adaptive capabilities of the immune system. 230 Uncommonly, this results in a chronic suppurative process, variants of which have been reported as ‘coral reef granuloma’ and blastomycosis-like pyoderma (see p. 553). Actinic comedonal plaque, in which fibrous tissue and comedones are present with some residual elastosis at the periphery, can be the end-stage picture of this inflammatory process.
Another secondary change that may occur in sun-damaged skin is the formation of actinic granulomas in which there is a granulomatous response to solar elastotic material and its resorption by macrophages and giant cells (elastophagocytosis, elastoclasis).231. and 232. Actinic granulomas present clinically as one or more annular lesions with an atrophic center and an elevated border. They are considered with the granulomatous tissue reaction (see p. 188). Elastophagocytosis has also been reported in association with various inflammatory processes in sun-protected skin. 233
Ultraviolet light is usually incriminated in the etiology of the degenerative changes. 234 Human studies have demonstrated that small amounts of UVA or solar-simulated UV are capable of producing cutaneous photodamage.235. and 236. Long-term exposure to PUVA (psoralens and ultraviolet A) is associated with persistent increases in actinic degeneration and pigmentary abnormalities on both sun-exposed and sun-protected sites. 237 However, it has been suggested that infrared radiation may also contribute, as changes characteristic of solar elastosis are seen in erythema ab igne. 238 Although not usually regarded as one of the elastotic syndromes, this condition will be considered in this section because of its similar histological appearances. Two further causes of heightened elastosis are cigarette smoking239.240. and 241. and photosensitivity resulting from the therapeutic use of hydroxyurea. 242 The degree of aging in photoprotected skin correlates significantly with patient age and a history of cigarette smoking. 243
The usual clinical appearance of solar elastosis is thickened, dry, coarsely wrinkled skin with loss of skin tone. 244 Sometimes there is a yellowish hue. There may be some telangiectasia and pigmentary changes (poikilodermatous changes) in severe cases.245. and 246. The best recognized clinical variant is cutis rhomboidalis in which there is thickened, deeply fissured skin on the back of the neck. Other clinical patterns include citrine skin, 247 Dubreuilh’s and other elastomas, 248 and solar elastotic bands of the forearm.249. and 250. Elastotic bands are characterized by cordlike plaques across the flexor surfaces of the forearms. 251 The author has seen a similar, but much milder phenomenon on the face of a colleague with severe solar damage. Bullous lesions are extremely rare.252. and 253.
The origin of the elastotic material has been the subject of much debate. It has been attributed to the degradation of collagen or elastic fibers or both.254. and 255. Alternatively, it has been suggested by others that the material results from the actinic stimulation of fibroblasts. 256 More recent work indicates that the elastotic material is primarily derived from elastic fibers.257. and 258. The increased elastin appears to result from transcriptional activation of the elastin gene.259. and 260. In contrast, various studies have clearly shown that in unexposed skin, elastin content decreases with age (−44%) between 50 and 70 years of age. Interestingly, the elastin content of moderately sun-exposed areas does not change during aging, which may be the result of age-induced reduction and sun-dependent increase in elastin. 261 In addition to the increased production of elastin in sun-exposed skin, there is reduced human leukocyte elastase activity, possibly a consequence of increased lysozyme production which protects elastin from proteolysis. 261 In contrast, neutrophil elastase is increased following experimental radiation to sun-protected skin. 262 Clusterin, a glycoprotein, is markedly increased in solar elastosis. Its role is not completely elucidated. 263 A small amount of type I and VI collagen and procollagen type III are present, but the significance of this finding remains uncertain. 257 DNA photodamage and UV-generated reactive oxygen species are the initial molecular events that lead to most of the typical histological and clinical manifestations of chronic photodamage of the skin. 264 Photoaging results in the accumulation of glycosaminoglycans on the elastotic material in the upper dermis and not between collagen and elastic fibers as in normal skin.265. and 266. Fibrillin is also increased. 258 Collagen VII is reduced and this may contribute to the formation of wrinkles by weakening the bond between the dermis and the epidermis. 267 D-aspartyl residues are increased in chronological and photo-induced aging. 219 Matrix metalloproteinase-7 and -12 are increased in photodamaged skin; they may contribute to remodeling of elastotic areas.268. and 269. Metalloproteinase-1 may be responsible for the degeneration and reduction in collagen.270.271. and 272. Fibulin-2, which belongs to a novel family of extracellular matrix protein, is significantly increased in actinically damaged skin. 11 Other consequences of photoaging are mutations in p53 and the partial loss of the ability of epidermal cells to differentiate normally. 273 The changes are qualitatively quite different from those seen in chronological aging,274. and 275. contrary to the assertion of some. 276
Various therapies have been used in recent times to improve the clinical appearance of photoaged skin. 226 One such technique, dermabrasion, produces clinical improvement by the synthesis of type I collagen. 277 Another technique, the prolonged application of topical tretinoin (retinoic acid), produces epidermal thickening,225.278.279.280. and 281. hypergranulosis, an increase in epidermal Langerhans cells, 231 the deposition of collagen in the papillary dermis, 282 and, sometimes, an increase in fine elastic fibers in the papillary dermis. 283 Carbon dioxide laser resurfacing, and the intradermal injection of crosslinked hyaluronic acid may also be used.226. and 284. These changes may result in an improved clinical appearance,285. and 286. although this has not occurred in all studies. 287 Imiquimod, tacrolimus ointment, topical 5-aminolevulinic acid, and topical estrogen creams have all been used successfully in the treatment of photoaging.288.289.290. and 291. Over-the-counter preparations have had variable results. 292 The prolonged use of sunscreens results in a significant reduction in the amount of solar elastosis and other harmful effects;293. and 294. it is the single most cost-effective therapy.225. and 266.
In mild actinic damage there is a proliferation of elastic fibers in the papillary dermis. These are normal or slightly increased in thickness. In established cases the papillary and upper reticular dermis is replaced by accumulations of thickened, curled, and serpiginous fibers forming tangled masses which are basophilic in hematoxylin and eosin-stained sections (Fig. 12.12). 22 Sometimes there are amorphous masses of elastotic material in which the outline of fibers is lost except at the periphery. These masses are thought to form from the tangled fibers, as transitions can be seen on electron microscopy. A thin grenz zone of normal-appearing collagen is present in the subepidermal zone. 295 This may have lost its network of fine vertical fibers. Collagen is reduced in amount in the reticular dermis. Telangiectases may be seen. 266 In a study of Korean skin with chronic photodamage, there was a gradual decrease in the number and size of dermal vessels over several decades of sun exposure. 296 Transepidermal elimination of elastotic material can occur. 297 This process is not uncommon following cryotherapy to severely damaged skin, which seems to trigger it in some individuals (Fig. 12.13). The elastotic material stains black with the Verhoeff stain (Fig. 12.14). Sometimes the homogeneous deposits are less well stained. Melanocytes and Merkel cells are both increased in number.298. and 299.
Solar elastosis with amorphous and fibrillary material in the upper dermis. (H & E)
Solar elastosis. Curled elastotic fibers are insinuating between basal keratinocytes. This represents the early stages of the transepidermal elimination of these damaged fibers. (H & E)
Perforating solar elastosis. The elastic fibers being eliminated are thick, curled, and serpiginous in morphology. (Verhoeff–van Gieson)
Biopsies from individuals with chronic sunlight exposure, some of whom had persistent erythema, have been described as showing a ‘perivenular histiocytic-lymphocytic infiltrate in which numerous mast cells, often in close apposition to fibroblasts, were observed’: this condition has been termed ‘chronic heliodermatitis’. 300 In normal-appearing sun-exposed skin there are more mast cells, macrophages, CD4+, CD45RO+ T cells and CD1a+ dendritic cells than sun-protected skin. 301
Epidermal changes also occur in severely damaged skin. The stratum corneum may be compact and laminated or gelatinous; it sometimes contains vesicles full of proteinaceous material. 302 In the malpighian layer, cell heterogeneity, vacuolization, and dysplasia may be found. 302
In bullous solar elastosis there is a well-defined, horizontally oriented cleft, lined by fibrin, in the middle of a dermis showing marked solar elastosis. 253 Red cell extravasation in the region of the cleft is not uncommon.
A spectrum of ultrastructural changes is found which parallels the clinical degree of damage.22.303. and 304. In mild cases, the elastic fibers in the papillary dermis are increased in number. The microfibrillar dense zones become irregular in outline, more electron dense, and many times larger. In severe cases the elastin matrix becomes granular and develops lucent areas around the microfibrillar dense zones. 22 Some fibers become disrupted and show a moth-eaten appearance or become transformed into finely granular bodies. 22 Similar ultrastructural findings have been reported in chronic radiodermatitis. 305 Deformed collagen fibers, of various diameters, are found in the papillary dermis. 306 Following PUVA therapy, the elastic fiber changes include a breakdown of the microfibrils and subsequent fragmentation of the elastic fibers. 307 Melanocytes show degenerative changes with the development of large intracytoplasmic vacuoles. 298
Scanning electron microscopy of solar elastosis shows some normal fibers, some thick damaged cylindrical fibers, and large masses of markedly changed fibers, which probably correspond to the amorphous deposits seen in severe cases. 308
The solar degenerative condition, nodular elastosis with cysts and comedones, is also known as the Favre–Racouchot syndrome. 309 It occurs as thickened yellowish plaques studded with cysts and open comedones.310.311. and 312. It involves the head and neck, but particularly the skin around the eyes. Lesions may extend to the temporal and zygomatic areas. A case involving the shoulder region has been reported. 313 Rare cases are unilateral. 314 There is a predilection for older males who have a history of prolonged solar exposure. Smoking may act in conjunction with solar damage to potentiate the development of this condition.315. and 316. A case precipitated by radiation therapy and successfully treated with low-dose isotretinoin has been reported. 317 Another case was provoked by ultraviolet (UVA1 and UVB) radiation. 318
In addition to the marked solar elastosis, there are dilated follicles and comedones which contain keratinous debris in the lumen. The sebaceous glands are often atrophic. A recent study of patients without much solar exposure showed multiple comedones without significant solar elastosis, suggesting that the two processes might be independent. 319
Elastotic nodules are small, usually asymptomatic, pale papules and nodules found predominantly on the anterior crus of the antihelix in response to actinic damage.320.321. and 322. They are often bilateral. There is a marked predilection for elderly white males. Rare cases develop on the helix, where they may be painful, simulating chondrodermatitis nodularis helicis. They may be diagnosed clinically as basal cell carcinoma, amyloid, or even small gouty tophi.
There is marked elastotic degeneration of the dermis with the formation of irregular, coarse elastotic fibers and larger clumped masses of elastotic material (Fig. 12.15 and Fig. 12.16). These changes are best seen with the Verhoeff elastic stain. The overlying epidermis shows mild to moderate orthokeratosis and some irregular acanthosis. There is mild telangiectasia of vessels in the papillary dermis, and some new collagen is often present in this area.
(A) Elastotic nodule of the ear. (B) Clumped masses of elastotic material are present in the mid dermis. (H & E)
Elastotic nodule of the ear. There are clumped masses of elastotic material. (Verhoeff–van Gieson)
Also known as ‘degenerative collagenous plaques of the hands’, ‘keratoelastoidosis marginalis’, and ‘digital papular calcific elastosis’, 323 collagenous and elastotic plaques of the hands is a slowly progressive, degenerative condition found predominantly in older males.324.325.326. and 327. There are waxy, linear plaques at the juncture of palmar and dorsal skin of the hands. The condition particularly involves the medial aspect of the thumbs and the lateral (radial) aspect of the adjacent index finger. In this respect the lesions resemble in part those seen in the genodermatosis acrokeratoelastoidosis (see p. 261). 324 Physical trauma of a repetitive nature and prolonged actinic exposure may play a role in the etiology of collagenous and elastotic plaques of the hand.326. and 328.
The most noticeable changes are in the dermis where there are numerous thick collagen bundles having a haphazard arrangement, but with a proportion running perpendicular to the surface (Fig. 12.17). 329 There is often a slight basophilic tint to the dermis; elastotic fibers can be seen in the lower papillary dermis and intimately admixed with the collagen bundles in the reticular dermis. Basophilic elastotic masses are found in the upper dermis. 323 The dermis shows reduced cellularity with large areas devoid of fibroblasts. 327 Sweat ducts may be mildly dilated in the mid dermis and compressed in other areas.
(A) Collagenous and elastotic plaque. (B) The collagen bundles in the upper dermis have a characteristic haphazard arrangement with some bundles arranged vertically. Elastotic material is admixed. (H & E)
In elastic tissue stains, the elastotic material in the lower papillary dermis is confirmed (Fig. 12.18). In the reticular dermis, granular and elastotic material can be seen in an intimate relationship within some of the larger collagen bundles. 324 In some cases, there are focal deposits of calcification in the dermis. The changes are distinct from those of solar elastosis.
Collagenous and elastotic plaque. Elastic tissue is reduced between the thickened vertical collagen. There are curled fibers in the papillary dermis. (Verhoeff–van Gieson)
The overlying epidermis may show mild hyperkeratosis and thickening of the granular layer. In some cases there is slight acanthosis, while in others there may be loss of the rete pattern.
Erythema ab igne refers to the development of persistent areas of reticular erythema, with or without pigmentation, at the sites of repeated exposure to heat, usually from open hearths.330. and 331. Other cases have included frequent hot bathing, 332 a laptop computer placed on the thighs, 333 and a heating pad. 334 The lower legs are usually involved. Erythema ab igne is now seen only rarely. 335 Keratoses and, rarely, squamous cell carcinomas may develop in lesions of long standing. 336
There is thinning of the epidermis with effacement of the rete ridges and some basal vacuolar changes. Areas of epithelial atypia, resembling that seen in actinic keratoses, are sometimes present. There is usually prominent elastotic material in the mid dermis. 338 A few large histiocytes may be present. A small amount of hemosiderin and melanin may be present in the upper dermis. 330
There are several distinct levels in the biosynthesis of elastic fibers at which errors can be introduced. These can lead to reduced production of elastic fibers or to the appearance of abnormal ones. Breakdown of fibers (elastolysis) is another mechanism which can lead to a reduction in the elastic tissue content of the dermis. This probably results from increased elastase activity.
The reduction in dermal elastic tissue can be generalized, as in cutis laxa, or localized, as in anetoderma and blepharochalasis. Cases with features intermediate between these two types or with fine wrinkling of the skin occur. Sometimes the reduction in elastic fibers is subclinical or overshadowed by other features. This is the case in various granulomatous inflammatory disorders.
Skin atrophy, associated with a decrease in elastic fibers, has been reported at the site of thiocolchicoside injections. 339
Nevus anelasticus is the term suggested by Staricco and Mehregan66 for several cases reported in the earlier literature characterized by an absence or definite reduction and/or fragmentation of elastic fibers in cutaneous lesions of early onset. 340 Further cases have been reported in which multiple papular lesions have developed, particularly on the trunk.340.341.342. and 343. The term papular elastorrhexis has been used for such cases; it has been regarded by some as a distinct variant of connective tissue nevus, 344 and not synonymous with nevus anelasticus. The author has been criticized in a recent paper for not separating cases of papular elastorrhexis from nevus anelasticus. 345 However, another recent paper by Ryder and Antaya has suggested that nevus anelasticus, papular elastorrhexis, and eruptive collagenoma are the same entity; their preference for a name for this composite entity was papular elastorrhexis. 346 The lesions are not perifollicular in distribution. Separation from the non-inflammatory type of anetoderma may be difficult (see below).
Sections show a localized reduction in elastic fibers, with normal collagen. 341 The elastic fibers may show intense fragmentation in some cases. 340 Fibers in the papillary dermis may be normal. There is no inflammation in nevus anelasticus, but mild dermal inflammation has been reported in papular elastorrhexis. 347 However this is a tenuous feature to use in separating these conditions as various elastic tissue diseases may have (mild) inflammatory stages.
Perifollicular elastolysis is a not uncommon condition of the face and upper back in which 1–3 mm gray or white, finely wrinkled lesions develop in association with a central hair follicle.348. and 349. Balloon-like bulging of larger lesions may develop. 348 The disorder is significantly associated with acne vulgaris.349. and 350. An elastase-producing strain of Staphylococcus epidermidis was found in the hair follicles located within lesions in one report. 348
There is an almost complete loss of elastic fibers confined to the immediate vicinity of hair follicles (Fig. 12.19). There is no inflammation.
Perifollicular elastolysis. There is absence of elastic tissue around the pilosebaceous follicle. A few thin fibers are present near the edge of the photomicrograph. (Verhoeff–van Gieson)
Anetoderma (macular atrophy) is a rare cutaneous disorder in which multiple, oval lesions with a wrinkled surface develop progressively over many years.351. and 352. Individual lesions may bulge outwards or be slightly depressed. They usually herniate inwards with finger tip pressure. There is a predilection for the upper trunk and upper arms, but the neck and thighs may also be involved. Facial involvement may lead to chalazodermia. 351 Onset of the lesions is in late adolescence and early adult life. Childhood cases have been reported. 353 Familial cases are quite rare.354.355.356.357.358. and 359.
The onset of lesions may be preceded by an inflammatory stage with erythematous macules and papules (Jadassohn–Pellizzari type) or there may be no identifiable precursor inflammatory lesions (Schweninger–Buzzi type). 352 These two types have been classified as primary anetodermas. As an inflammatory infiltrate may be present even in cases with no clinical inflammatory features, this classification is outdated. 360 Patients with primary anetoderma frequently have at least one prothrombotic abnormality, the most common being the presence of antiphospholipid antibodies.360.361.362. and 363.Secondary anetodermas351. and 352. develop during the course of other diseases such as syphilis, leprosy, sarcoidosis, granuloma annulare, 364 tuberculosis, varicella, 365 HIV infection,366. and 367. folliculitis, 368 angular cheilitis, 369 Lyme disease, 370 acrodermatitis chronica atrophicans, 351 lupus erythematosus, 351 amyloidosis, lymphocytoma cutis, 371 cutaneous B-cell lymphoma,372. and 373. mycosis fungoides, 374 juvenile xanthogranuloma,375. and 376. immunocytoma, 377 or following penicillamine therapy378 or hepatitis B immunization. 379 Patches of anetoderma may develop in extremely premature infants, 380 usually at the sites of attachment of gel electrocardiographic electrodes.381. and 382. Anetoderma-like changes have been reported in a patient with the clinical features of atrophoderma. The term ‘atrophoderma elastolytica discreta’ was used for these lesions. 383 The association with urticaria pigmentosa may be coincidental. 384 Rarely, secondary anetoderma overlies a pilomatrixoma.385. and 386. The lesions of secondary anetoderma do not always correspond with those of the primary disease process.
A variety of ocular and skeletal defects have been reported in individuals with anetoderma. They have been chronicled in a review of the extensive European literature on this condition. 351
Theoretically, anetoderma could result from increased degradation or reduced synthesis of elastic tissue. 387 It has been suggested that all cases have an inflammatory pathogenesis, which would tend to indicate that an elastolytic process is operative.21. and 388. Increased expression of various metalloproteinases has been reported; they play an important role in the degradation of elastic fibers in anetodermic skin.19. and 389. The concentration of elastin, as measured by the desmosine content of the skin, is markedly reduced. 387 Immunological abnormalities, the most common of which is a positive antinuclear factor, have been documented.390.391. and 392.
If a biopsy is taken from a clinically inflammatory lesion, the dermis will show a moderately heavy perivascular and even interstitial infiltrate, predominantly of lymphocytes. Plasma cells and eosinophils are occasionally present. Neutrophils have been noted sometimes in very early lesions. 388
In established lesions, most reports have noted an essentially normal appearance in hematoxylin and eosin-stained sections. However, in one large series, a perivascular infiltrate of lymphocytes was found in all cases. 388 There was a predominance of helper T cells. 393 The authors of that account did not attempt to reconcile their findings with earlier reports in which inflammatory cells were noted to be absent.352.387. and 388.
Scattered macrophages and giant cells, some showing elastophagocytosis, may also be present. 394 Non-caseating granulomas were present in one case, in association with Takayasu’s arteritis. 395
Elastic tissue stains show a normal complement of fibers in the early inflammatory lesions. In established lesions, elastic fibers are sparse in the superficial dermis and almost completely absent in the mid dermis (Fig. 12.20).
Anetoderma. There is a heavy infiltrate of lymphocytes in the mid dermis. Beneath this the elastic fibers have almost completely disappeared. Five years after this biopsy was taken the patient developed cutaneous lupus erythematosus. (Verhoeff–van Gieson)
Direct immunofluorescence in some cases of primary anetoderma shows a pattern of immune deposits similar to that of lupus erythematosus. 396 There are no other manifestations of the latter disease in these cases.
The term ‘cutis laxa’ encompasses a group of rare disorders of elastic tissue in which the skin hangs in loose folds, giving the appearance of premature aging.347. and 399. In many cases, there is a more generalized loss of elastic fibers involving the lungs, gastrointestinal tract and aorta, leading to emphysema, hernias, diverticula, and aneurysms.400.401.402. and 403. It is an etiologically heterogeneous disorder.
Congenital and acquired forms exist. Congenital cutis laxa is genetically heterogeneous: there are several different autosomal recessive forms of the disease (OMIM 219100), one of which is associated with growth retardation.404.405.406.407. and 408. These cases were reported in the earlier literature and may be examples of the De Barsy syndrome (see below). There is an autosomal dominant form (OMIM 123700) which is less severe. 409 One of the dominant forms is allelic to the Williams–Beuren syndrome and is related to mutations in the elastin (ELN) gene. 410 The X-linked recessive variant (OMIM 304150), 411 in which there is a deficiency of lysyl oxidase, is now regarded as a variant of Menkes’ syndrome (OMIM 309400);410.412. and 413. the two are allelic. 410 It is caused by mutations in the ATP7A gene. 414 The congenital forms are associated with a characteristic facies, with a hooked nose and a long upper lip (‘blood-hound’ facies). 415 Congenital cutis laxa has been reported in a young male with the Kabuki make-up syndrome.416. and 417. It has also been reported in a newborn with congenital hypothyroidism due to isolated thyrotropin deficiency. 418
More than 50 cases of acquired cutis laxa have been described. The changes may be generalized or localized.419.420.421.422.423. and 424. Localized cases may be acral or cephalic in distribution. 425 Acquired cutis laxa may be of insidious onset426 or develop after a prior inflammatory lesion of the skin427 which may take the form of erythema, erythema multiforme, urticaria,428. and 429. a vesicular eruption, including dermatitis herpetiformis, 430 or Sweet’s syndrome. 431 Several cases have followed an allergic reaction to penicillin,432. and 433. while others have been associated with isoniazid therapy, 434 mastocytosis, 435 myelomatosis,436.437.438. and 439. cutaneous lymphoma,440. and 441. rheumatoid arthritis, 442 systemic lupus erythematosus, 443 or the nephrotic syndrome. 399 In a case associated with celiac disease, deposits of IgA were present on the dermal elastic fibers. 444 Cutis laxa may occur as a manifestation of pseudoxanthoma elasticum. Late-onset cutis laxa also occurs in hereditary gelsolin amyloidosis (type V – familial amyloidosis of the Finnish type (OMIM 105120)) caused by a G654A or G654T gelsolin gene mutation on chromosome 9q34.445. and 446. Gelsolin amyloid is deposited in dermal nerves, blood vessels, and appendages, and often encircles elastic fibers in the lower dermis leading to fragmentation and loss of fibers. 445
The congenital cases result from a defect in the synthesis or assembly of the components of the elastic fiber. All patients with the autosomal dominant form of cutis laxa who have had genetic sequencing studies performed have had mutations in the ELN gene localized to chromosome 7q11.2. 447 Its product, tropoelastin, is the most abundant component of elastic fibers.414. and 448. Mutations in the ELN gene not only cause cutis laxa448 but also cause subvalvular aortic stenosis, 449 which may occur as an isolated disease or as part of the Williams–Beuren syndrome (Williams syndrome), a microdeletional syndrome that involves the deletion of one complete copy of ELN (see below).414. and 450. The De Barsy syndrome appears to be another subgroup of cutis laxa that may involve a defect in elastin production with increased degradation. 451 This syndrome is also characterized by progeroid features and mental retardation. 452
The molecular defects underlying the recessive forms of cutis laxa have recently been discovered.8. and 9. Mutations in the fibulin-5 gene (FBLN5) were the first identified. 410 Mutations in this gene are also responsible for age-related macular degeneration, the leading cause of irreversible visual loss in the Western world. 453 A missense mutation in the fibulin-4 (FBLN4) gene has also resulted in cutis laxa. 12 A congenital disorder of glycosylation involving a defect in the biosynthesis of N- and O-glycans has also been found in patients with cutis laxa. 17 Other rare syndromes associated with cutis laxa in which the mechanism of the decrease in fibers is not known include the Barber–Say syndrome (OMIM 209885), characterized by a dysmorphic face and hypertrichosis, 454 the SCARF syndrome (OMIM 312830), with skeletal abnormalities, 455 and geroderma osteodysplasticum (OMIM 231070). 456 It has been suggested recently that the elastic tissue abnormality in geroderma osteodysplasticum has overlap features between cutis laxa and wrinkly skin syndrome (OMIM 278250). 457
In those acquired cases associated with severe dermal inflammation, it has been suggested that granulocytic elastase may be responsible for the degradation of the elastic fibers. Cultured fibroblasts from one case showed increased elastase activity. 429 One report suggested that several factors, including high levels of cathepsin G, low lysyl oxidase activity, and a reduction in circulating proteinase inhibitor(s), could all contribute to the loss of elastin. 458 Collagenase and gelatinase A and B expression is up-regulated at the transcriptional level in cutis laxa. 459 This may explain the collagen abnormalities (see below) that are sometimes found. 460 A more recent paper has described missense alleles in both the elastin and fibulin-5 genes in a patient with acquired cutis laxa with inflammatory destruction of elastic fibers, suggesting an underlying genetic susceptibility in some patients with acquired cutis laxa. 461
Localized areas of loose skin may develop in cutaneous lesions of sarcoidosis, syphilis, and neurofibromatosis. 426 Loose skin localized to the hands, feet, and neck, and deep palmar and plantar creases are seen in Costello syndrome (OMIM 218040) in which there are also characteristic facies, mental retardation, growth disorders, and, sometimes, hypertrophic cardiomyopathy.462.463. and 464. Acanthosis nigricans may also be present. 465 There is an increased susceptibility to bladder cancers and rhabdomyosarcoma. It results from a functional deficiency in elastin-binding protein (EBP).7. and 462. Mutations in the HRAS gene on chromosome 11p15.5 account for the majority of cases of Costello syndrome but other candidate genes have been mentioned sporadically.
The fine elastic fibers in the papillary dermis are lost and there is a decrease in fibers elsewhere in the dermis (Fig. 12.21). Remaining fibers are often shortened and they vary greatly in diameter. The borders are sometimes indistinct and hazy. Fragmentation of fibers may be noted. Giant cells are rarely present, phagocytosing elastic fibers. A variable inflammatory reaction is present in the acquired cases with an associated clinical inflammatory component. 428 In several cases, the inflammatory infiltrate has been quite heavy, with neutrophils, eosinophils, and lymphocytes in the superficial and deep dermis. 433 Deposits of immunoglobulins have been demonstrated on elastic fibers in the dermis in several cases.439. and 444.
(A) Cutis laxa. (B) There is an almost complete absence of elastic fibers. (Orcein)
Shortening and rupture of elastic fibers are seen in Costello syndrome. There are decreased amounts of elastin. 463
The elastic tissue varies in content, appearance, and the proportion and manner by which elastin and the microfibrillar component associate.468. and 469. The microfibrils are reduced in the papillary dermis. 470 There is some fragmentation of elastic fibers with accumulation of granular material. 436 Fragmented fibers are sometimes surrounded by fibroblasts or macrophages. Abnormalities of collagen structure have been noted in a few reports,468. and 471. but specifically excluded in others. 436 An unusual case of acquired cutis laxa, associated with the cutaneous and systemic deposition of a fibrillar protein, has been reported. 472
Elastolysis of the earlobes may represent a variant of cutis laxa confined to the earlobes. 473 This is supported by cases with associated facial involvement.429. and 473.Blepharochalasis is a similar condition that presents with recurrent bouts of painless edema of the eyelids and periorbital region resulting in degradation of elastic fibers and their subsequent loss from the dermis.443. and 474. The floppy eyelid syndrome is morphologically similar. 475
Williams–Beuren syndrome (OMIM 194050), also known as Williams’ syndrome, is a multisystem, congenital disorder characterized by craniofacial, neurobehavioral, cardiovascular, and metabolic changes.450. and 476. It results from a microdeletion in the q11.23 region of chromosome 7, involving the elastin gene and up to 26 other genes such as the LIMK1 gene. 477 It is therefore a contiguous gene syndrome. Genes on other chromosomes have also been implicated. Its prevalence is approximately 1 in 10 000 births. Despite a moderate reduction in elastin deposition in the skin, the clinical changes are relatively mild with increased softness and mobility of the skin. 476
The overall appearance of the skin in H & E sections is normal. Morphometric analyses of elastic fibers have demonstrated a marked reduction in elastic fiber diameter and volume compared with healthy controls. 65
Papillary-dermal elastolysis is a rare disorder of elastic tissue characterized by clinical lesions resembling pseudoxanthoma elasticum, with small papules and cobblestone plaques on the neck and upper trunk.25. and 479. Reported cases have occurred exclusively in females, but if the merged entity (see below) is accepted, both sexes may be involved. 480 Similar histopathological changes were present in a case presenting as a small, hyperpigmented plaque. 481 It has been suggested recently that this condition is part of the spectrum of white fibrous papulosis of the neck (see p. 317), for which the term ‘fibroelastolytic papulosis of the neck’ has been suggested. 480 The pathogenesis of these two merged entities is unknown; it is possibly related to intrinsic aging.480. and 482.
The coexistence of papillary-dermal elastolysis and linear focal dermal elastosis in the same patient has been reported. 483
There is a complete loss of elastic fibers in the papillary dermis. The remaining fibers are not calcified or fragmented; that is, there are no histopathological features of pseudoxanthoma elasticum. Elastophagocytosis was present in one case, suggesting that this may be the mechanism for the loss of elastic fibers. 484 Immunohistochemical studies have demonstrated a disappearance of both elastin and fibrillin-1 from the papillary dermis, suggesting that this condition is more than an age-related state.485. and 486.
Mid-dermal elastolysis, first described by Shelley and Wood in 1977, 487 is characterized by widespread patches of fine wrinkling due to a loss of elastic fibers from the mid dermis.487.488.489.490.491. and 492. The clinical features can be quite subtle. 493 A few cases have shown a second clinical feature with looseness of the skin around hair follicles. 494 Other cases have had flesh-colored papules in a perifollicular distribution. 495 A third clinical pattern with a prominent reticular appearance has been reported. 496 It may represent the end stage of granuloma annulare. 496 Most cases have involved the upper extremities, neck, and trunk of women. It may represent a variant of anetoderma.
In nearly 50% of cases, erythema, urticaria, or burning precedes or coincides with the development of the lesions, suggesting that an inflammatory process may be involved in the pathogenesis. In some cases, the condition appears to be photoinduced or photoaggravated.495.497. and 498. The onset has followed augmentation mammoplasty with silicone implants, 499 granuloma annulare,500. and 501. and lupus erythematosus. 502 Lesions may remain stable for many years. Mid-dermal elastolysis has been reported in one family with the Keutel syndrome (OMIM 245150), a rare autosomal recessive syndrome, characterized by abnormal cartilage calcification and due to mutations in the matrix Gla protein (MGP) gene. 503
There is some similarity to the cases reported from South Africa and South America, in young children, in whom wrinkling developed after a preceding inflammatory stage.504.505. and 506.
An immunohistochemical study has shown enhanced expression of CD34+ and CD68+ cells and of matrix metalloproteinase-1 (MMP-1). 507 Another study confirmed the increase in CD68+ cells, but found that MMP-9 was present in epidermal keratinocytes and in large cells in lesional dermis. 495 This study of 11 patients concluded that the onset of the disease is strongly associated with UV exposure, which may induce fibroblast-like cells to express MMP-9 that in turn could be involved in the degradation of elastic fibers. 495 MMP-9 was also implicated in another study, in the destruction of the elastic fibers in this condition. 193
Sections stained with hematoxylin and eosin may appear normal, although in the early inflammatory stage a mild infiltrate of lymphocytes is present around vessels and, to a lesser extent, in interstitial areas.494. and 508. Spindle-shaped cells and large multinucleated cells with angulated outline are scattered between collagen bundles in the mid dermis in these early lesions. 495 They did not stain for CD68 or factor XIIIa in one study. 495 Phagocytosis of elastic fibers has been present in some cases,491. and 509. but specifically excluded in others.494. and 497. This may, of course, be related to the age of the lesion biopsied. Two cases of mid-dermal elastophagocytosis, presenting as persistent reticulate erythema, have been reported. 510
There is one report of a patient with mid-dermal elastolysis in which the initial erythematous and urticarial plaques revealed a neutrophilic infiltrate in the papillary and mid dermis and a normal pattern of elastic fibers. 511 In addition to the neutrophils there was leukocytoclastic debris, some endothelial swelling of vessels, and a few admixed lymphocytes, histiocytes, and eosinophils. 511
Stains for elastic tissue show an absence of fibers in the mid dermis. Elastic tissue is usually preserved around appendages, even in the clinical subset with perifollicular involvement. 512 There is no involvement of the papillary dermis or the lower reticular dermis. 494
Menkes’ kinky hair syndrome (OMIM 309400) is a rare multisystem disorder of elastic tissue transmitted as an X-linked recessive trait.513.514. and 515. The defective gene (ATP7A) has been localized to chromosome Xq12–q13.516. and 517. Characteristically the hair is white, sparse, brittle, and kinky. It looks and feels like steel wool. Pili torti and, occasionally, monilethrix are present. Neurodegenerative changes, vascular insufficiency, hypothermia, and susceptibility to infections are other manifestations of this syndrome.38. and 515. Mild forms occur. 518
The finding of reduced serum copper levels led to the view that Menkes’ syndrome was a simple copper deficiency state akin to that seen in copper-deficient sheep.519. and 520. It is now thought to be due to a spectrum of mutations in the copper-transporting ATPase gene, ATP7A. 7 There is reduced activity of the copper-dependent enzyme lysyl oxidase in fibroblasts derived from the skin of patients with this syndrome. 521 This enzyme is necessary for the crosslinking of elastin. 4 It has been suggested that this syndrome should be reclassified with Ehlers–Danlos syndrome type IX, in which a disorder of lysyl oxidase also occurs (see p. 328).
There are various hair shaft abnormalities which include pili torti, monilethrix, and trichorrhexis nodosa. 515 The internal elastic lamina of vessels is fragmented and there is intimal proliferation. Dermal elastic tissue appears to be unaffected.
The elastic fibers in the reticular dermis show a paucity of the central amorphous component while retaining normal microfibrillary material. 516
Fragile X syndrome (OMIM 300624), a rare X-linked form of mental retardation, is associated with a characteristic facies and connective tissue abnormalities which are clinically reminiscent of cutis laxa and the Ehlers–Danlos syndromes.522. and 523. Most cases result from an increase in length of a stretch of CGG triplet repeats in the FMR1 gene situated on the long arm of the X chromosome (Xq27.3).524.525. and 526. Rarely, the condition results from the deletion of all or part of this gene.525. and 527. Attention deficit hyperactivity disorder is a frequent behavioral problem in young boys with fragile X syndrome. 528
There is a reduction in dermal elastic tissue. The fibers are fragmented and curled and lack arborization. There is a reduction in stromal acid mucopolysaccharides.
Wrinkly skin syndrome (OMIM 278250) is a rare autosomal recessive disorder characterized by wrinkled skin with poor elasticity over the abdomen and on the dorsum of the hands and feet.529. and 530. It has some features overlapping with cutis laxa type II. 531 A recent study was unable to distinguish between wrinkly skin syndrome and cutis laxa with growth and developmental delay (OMIM 219200). 457 Increased palmar and plantar creases, a prominent venous pattern on the chest, microcephaly, and musculoskeletal abnormalities form part of the syndrome. There is overlap between wrinkly skin syndrome and geroderma osteodysplasticum (OMIM 231070).457. and 532.