Disorders of Cornification
Leonard Milstone M.D.
William Rizzo M.D.
Gabrielle Richard M.D.
Clinical Pearls
(LM)
(WR)
(GR)
Ichthyosis Vulgaris
Inheritance
Autosomal dominant; gene locus unknown
Prenatal Diagnosis
None
Incidence
1:250-1:2,000; M=F
Age at Presentation
Three months to 1 year of life
Pathogenesis
Retention hyperkeratosis with normal epidermal proliferation; defect in profilaggrin synthesis with subsequent decreased levels of profilaggrin in keratinocytes; most likely polygenic disease
Key Features
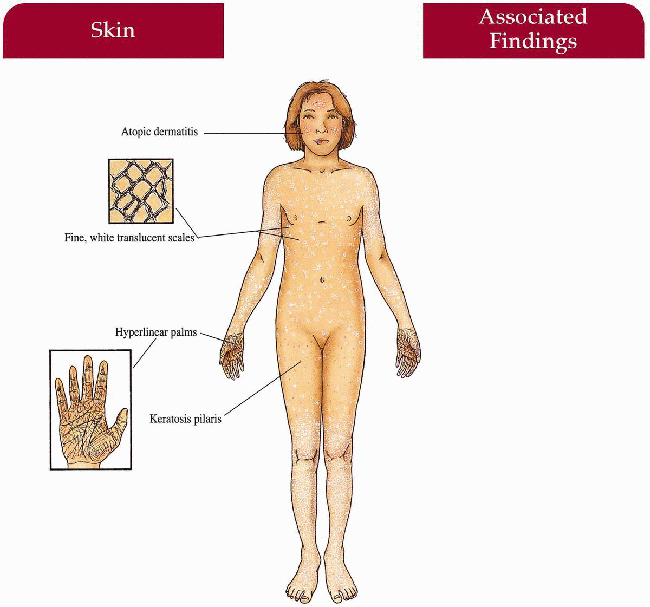
Skin
Fine, whitish, adherent scale sparing flexures with increased involvement on extensor extremities; face usually spared but may involve cheeks and forehead Atopic dermatitis (>50%)
Keratosis pilaris
Palmoplantar markings accentuated; rarely frank keratoderma
Differential Diagnosis
Atopic dermatitis
Xerosis
Acquired ichthyosis
X-linked ichthyosis (p. 4)
Lamellar ichthyosis (p. 10)
Laboratory Data
Skin biopsy from anterior shin—absent granular layer
Electron microscopy—small, poorly formed keratohyalin granules
Management
Referral to dermatologist—topical emollients
Prognosis
Improves in summer and with age; also improves in warm, moist environment
Clinical Pearls
This is a surprisingly difficult diagnosis to make with certainty—no genetic markers, no characteristic scale, and absent granular layer in only a small minority…Since the stratum corneum does not retain water well becaue of its inadequate endogenous humectant production, emollients containing urea or alpha-hydroxy acids work best. LM
|
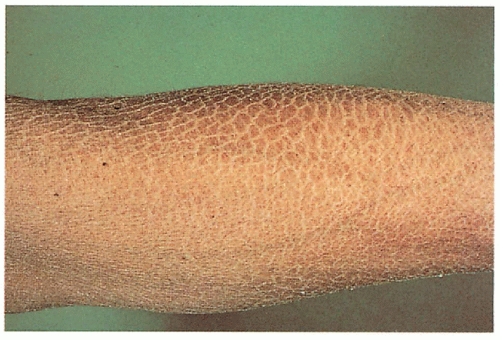
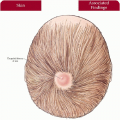
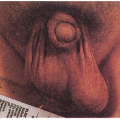
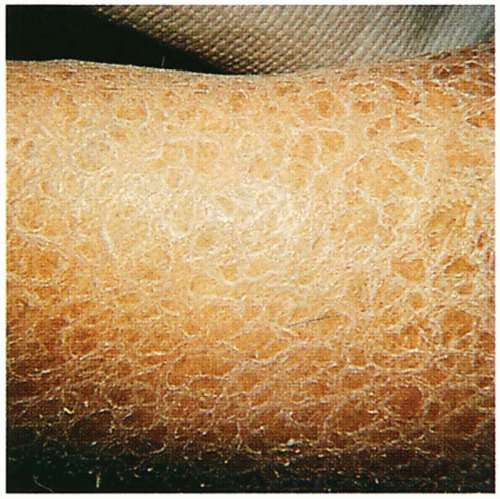 1.1. Translucent, adherent scale on extremity (1). |
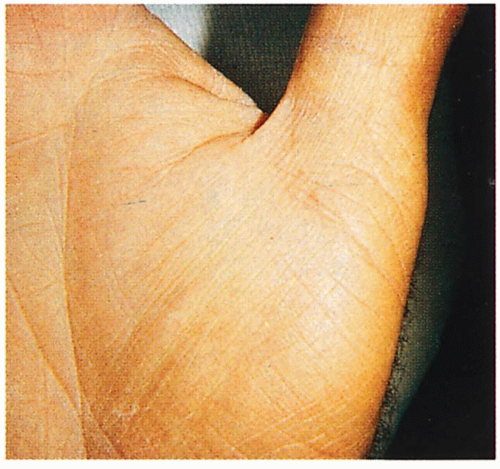 1.2. Accenuated palmar markings (1). |
X-linked Ichthyosis
Synonym
Steroid sulfatase deficiency
Inheritance
X-linked recessive: steroid sulfatase gene (STS) on Xp22.32
Gene deletions most common mutation (90%); contiguous gene deletion syndrome (10%)
Prenatal Diagnosis
Amniocentesis/chorionic villus sampling (CVS)—steroid sulfatase assay, increased dehydroepiandrosterone sulfate (DHEAS) levels
DNA analysis
Maternal estriol (serum/urine) and dehydroepiandrosterone levels
Incidence
1:2,000-1:6,000 males
Age at Presentation
Two to 6 weeks old
Pathogenesis
Steroid sulfatase gene deletion leads to decreased steroid sulfatase activity in stratum corneum with increased cholesterol sulfate and decreased cholesterol levels; may play a role in retention hyperkeratosis
Contiguous gene deletion syndrome may result in Kallmann syndrome and X-linked recessive chondrodysplasia punctata
Failure of labor to begin or progress in mother carrying affected fetus because of decreased placental sulfatase and estrogen and increased fetal DHEAS
Key Features
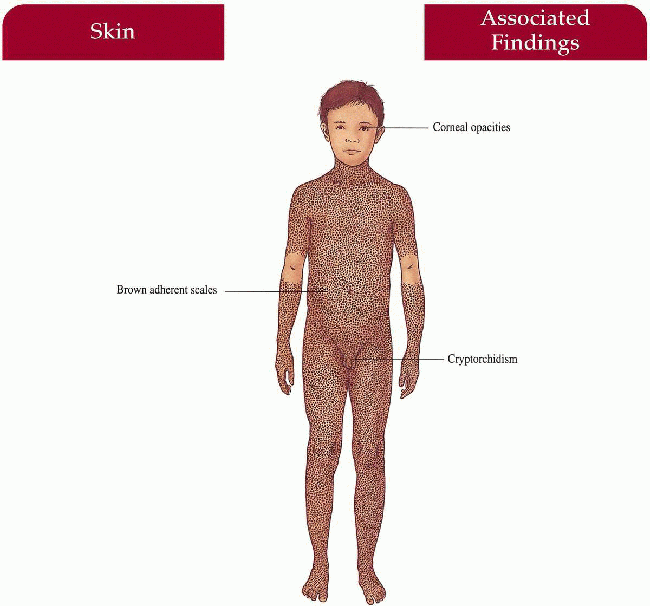
Skin
Brown, firmly adherent scale increased on extensors, posterior neck, trunk with relative sparing of flexures; sparing of palms, soles, face
Eyes
Comma-shaped corneal opacities—asymptomatic (50% of adult males, some female carriers)
Obstetrics
Placental sulfatase deficiency—failure of labor to begin or progress in mother carrying affected fetus
Genitourinary
Cryptorchidism (20%) with possible increase in testicular cancer
Differential Diagnosis
Ichthyosis vulgaris (p. 2)
Epidermolytic hyperkeratosis (p. 6)
Lamellar ichthyosis (p. 10)
Contiguous gene syndromes
Laboratory Data
Steroid sulfatase activity assay in scales, cultured fibroblasts, leukocytes
Lipoprotein electrophoresis—increased mobility of low-density lipoproteins
Serum cholesterol sulfate levels—increased
Management
Thorough physical examination by pediatrician
Referral to dermatologist—topical emollients
Referral to pediatric urologist if symptomatic
Advise obstetrician of potential complications
Prognosis
Cutaneous involvement waxes and wanes throughout life with seasonal variation
Clinical Pearls
A significant number of these cases are caused by chromosomal deletions…Indeed, most of my recent new cases have been referred to me after prenatal fluorescent in situ hybridization (FISH) screening…Such cases need follow-up for contiguous gene syndromes…All should be checked for undescended testes and risk for testicular carcinoma is increased even in absence of undescended testes…Scaling can be quite variable, even for an individual…Boys with extensive cradle cap at birth can have little/no scale at 1 year of age and then develop characteristic extensor scaling at 4 to 5 years of age…Small, tightly adherent scales on flanks often give a wrinkled or pseudoatrophic appearance…Water is critical for these folks…Some with marked scale in the dry winter have little or none during humid summers…I like alphahydroxy acids for RXLI. LM
|
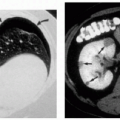
 1.3. Adherent, “dirty” brown scale (2). |
 1.4. Schematic diagram depicting comma-shaped corneal opacities (3). |
Epidermolytic Hyperkeratosis
Synonym
Bullous congenital ichthyosiform erythroderma
Bullous ichthyosis
Inheritance
Autosomal dominant; 50% spontaneous mutations; keratin K1, K10 genes on 12q, 17q respectively
Prenatal Diagnosis
Fetal skin biopsy at 20 to 22 weeks—clumped keratin filaments on electron microscopy
DNA analysis: K1 and K10 mutations if defect in family known, linkage analysis if kindred is large
Incidence
Rare—approximately 3,000 Americans afflicted; M=F
Age at Presentation
Birth
Pathogenesis
Heterogeneous gene defects in K1, K10 leads to defective keratin filaments in the upper epidermis with subsequent tonofilament clumping and bullae formation; arg res 156 of K10 is most common site for mutation with greatest severity at terminal rod regions
Extensive epidermal nevi (ichthyosis hystrix) reflect a somatic mosaicism for K1/K10 mutations; if gonadal mosaicism, then may have offspring with fullblown epidermolytic hyperkeratosis
Key Features
Skin
Newborn
Widespread bullae, erythroderma, denuded skin; secondary sepsis, electrolyte imbalance; ± focal areas of hyperkeratosis
Later Infancy to Adulthood
Localized to generalized hyperkeratosis with rare, focal bullae secondary to infection (Staphyloccus aureus, gram-negative bacteria); dark, warty scales with spiny ridges, increased in flexures; secondary bacterial infection with foul odor in macerated, intertriginous areas; scales shed with full-thickness stratum corneum leaving tender, denuded base; prominent palmoplantar keratoderma (in some patients); secondary nail dystrophy
Differential Diagnosis
Newborn
Epidermolysis bullosa (p. 200)
Staphylococcal scalded skin syndrome
Toxic epidermal necrolysis
Other causes of blistering
Later Infancy to Adulthood
Other ichthyoses
Laboratory Data
Skin biopsy for hematoxylin and eosin (H&E), frozen section (in newborn), and electron microscopy
Bacterial culture
Management
Newborn
Transfer to neonatal intensive care unit—monitor fluid, electrolytes, sepsis workup; intravenous (IV) broad-spectrum antibiotics until cultures negative; gentle handling, protective isolation
Later Infancy to Adulthood
Avoid topical keratolytics, salicylic acid, corticosteroids; systemic retinoids—short course in adulthood for flares; emolliation; antistaphylococcal, gram-negative antibiotic coverage; antibacterial soaps—Betadine, Chlorhexidine, Clorox in bath
Prognosis
Widespread blistering clears after newborn period; hyperkeratotic scale usually lifelong; generalized involvement may improve to localized disease after puberty
Clinical Pearls
Although scale is the major manifestation of this disease, this is a disease of keratinocyte fragility…Friction is the cause of most blisters, and repeated shear at body folds causes accentuated scale in those locations. Blisters in the absence of friction suggest Staphylococcus aureus…For most of my patients Aquaphor is their favorite topical…Strong topical keratolytics remove too much stratum corneum and leave the skin surface denuded and raw…ltching is unexplained, but can be severe…Oral retinoids are very effective for many, not all…Pregnancy and skeletal toxicity are ongoing concerns of oral retinoid use…Odor is a problem, somewhat reduced by hypochlorite (0.05% or 1:100 Clorox) and salt (2-3%) baths. Watch out for contractures, especially of palms, secondary to pain and/or blisters…Early referral to F.I.R.S.T., the patient support. LM
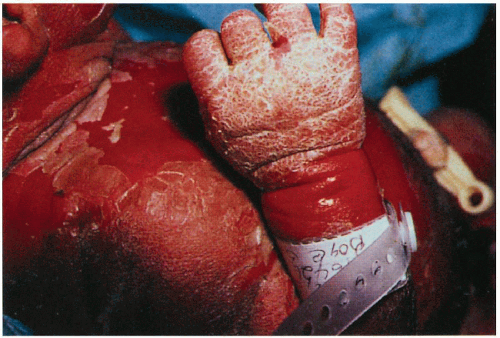 1.5. Infant with erythroderma, erosions, and hyperkeratosis (4). |
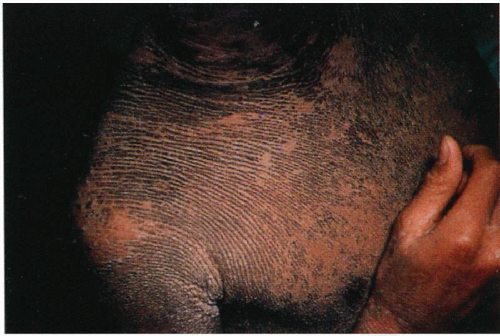 1.6. Adult with generalized hyperkeratosis. Note corrugated pattern to scale (1). |
|
Lamellar Ichthyosis
Inheritance
Autosomal recessive; transglutaminase 1 (TGM1) gene on 14q11
Prenatal Diagnosis
Chorionic villus sampling (CVS)/amniocentesis: TGM1 gene mutation or linkage analysis in families where molecular defect is known; fetal skin biopsy at 22 weeks
Incidence
Less than 1:300,000; M=F
Age at Presentation
Birth
Pathogenesis
Heterogeneous mutations in the TGM1 gene interfere with the normal cross-linking of structural proteins in the protein and lipid envelope of the upper epidermis leading to defective cornification and desquamation
Key Features
Skin
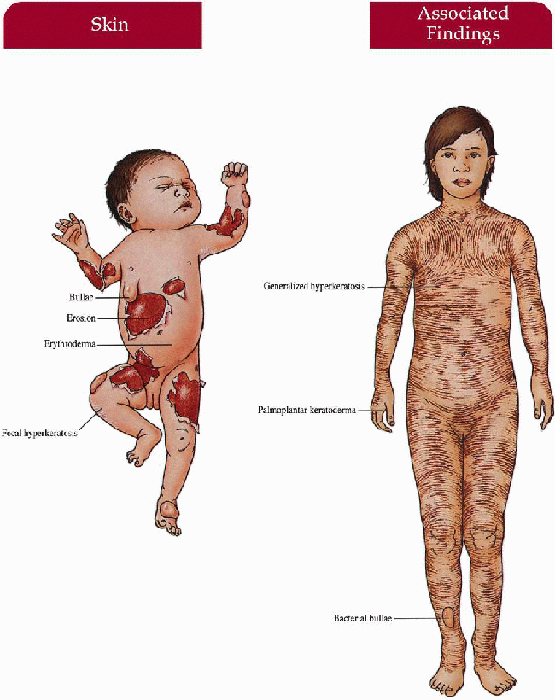
Newborn
Collodion baby with translucent membrane encasing body, ectropion, eclabium, generalized erythroderma; at risk for secondary sepsis, hypernatremic dehydration; membrane shed in first few days to weeks of life
Child/Adult
Generalized large, dark, platelike scale increased in flexures; erythroderma; ectropion; palmoplantar keratoderma; decreased sweating with heat intolerance
Hair
Scarring alopecia
Nails
Secondary dystrophy with nail fold inflammation
Differential Diagnosis
Epidermolytic hyperkeratosis (p. 6)
X-linked ichthyosis (p. 4)
Congenital ichthyosiform erythroderma (p. 12)
Netherton syndrome (p. 24)
Trichothiodystrophy (p. 246)
Laboratory Data
Skin biopsy-in situ detection of transglutaminase-1 expression and activity
Light microscopic hair examination (if alopecia)
Sepsis workup (newborn)
Management
Newborn
Transfer to neonatal intensive care unit—monitor fluids, electrolytes, and for sepsis; emolliation, high-humidity chamber
Child/Adult
Retinoids
Emolliation
Counsel regarding: avoiding strenuous activity, overheating
Prognosis
Severe involvement throughout life; normal life span
Clinical Pearls
Great variability in size and thickness of scales…Ectropion results from thick scale on the eyelids…I’ve had success using topical retinoids to reduce ectropion…but oral retinoids work better…Most patients try oral retinoids at one time or another, but retinoids unmask the underlying erythema and some patients would rather be scaly than red…Pregnancy and skeletal toxicity are ongoing concerns with retinoids…Educate patients about heat stroke and normal activity can be pursued…One of my patients ran crosscountry in college, another played lacrosse…Early F.I.R.S.T referral can be helpful. LM
|
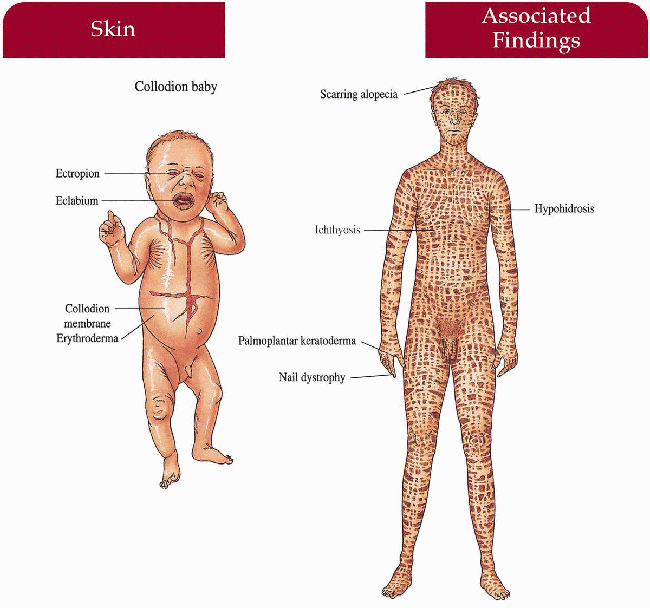
 1.7. Collodion baby with ectropion, eclabium (5). |
 1.8. Generalized scale on trunk and extremities (1). |
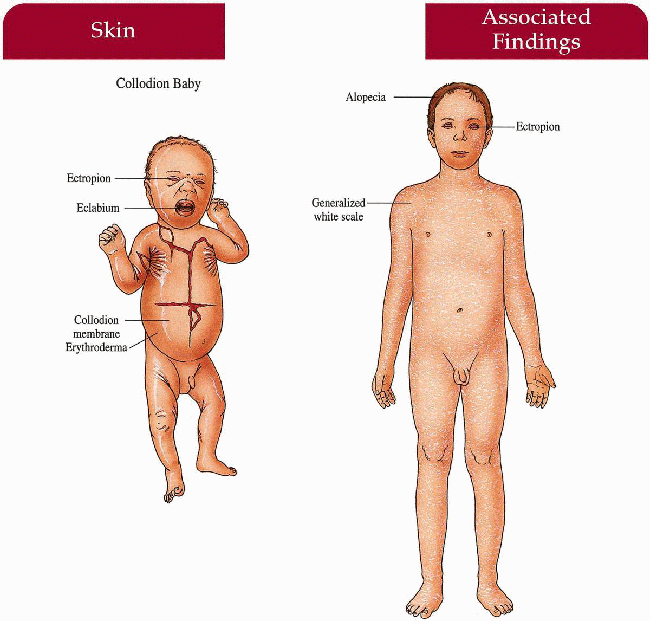
Congenital Ichthyosiform Erythroderma (CIE)
Synonym
Nonbullous CIE
Inheritance
Autosomal recessive; heterogeneous genetic loci
Prenatal Diagnosis
Fetal skin biopsy at 22 weeks
Incidence
1:180,000; more common than lamellar ichthyosis; M=F
Age at Presentation
Birth
Pathogenesis
TGM1 gene mutations have been identified in some patients (much more closely identified with lamellar ichthyosis); other gene loci have been linked as well; accelerated epidermal cell turnover rate
Key Features
Skin
After infancy
Generalized erythroderma with fine, white scale, flexures involved; extensor legs with large, platelike, dark scale; ± palmoplantar keratoderma; hypohidrosis with heat intolerance
Hair
Cicatricial alopecia
Eyes
Ectropion
Differential Diagnosis
Neutral lipid storage disease (NLSD)
Lamellar ichthyosis (p. 10)
Ichthyosis vulgaris (p. 2)
Netherton syndrome (p. 24)
Laboratory Data
Complete blood count (CBC)—differentiate from NLSD with lipid vacuoles in leukocytes and monocytes in NLSD
Skin biopsy—differentiate from NLSD with lipid vacuoles in basal epidermis in NLSD
Management
Newborn
Transfer to neonatal intensive care unit—monitor fluids, electrolytes, and for sepsis; emolliation, high-humidity chamber
Child/Adult
Topical keratolytics, topical retinoids, emolliation
Oral retinoids (short course)
Prognosis
Usually unremitting course but may improve at puberty
Clinical Pearls
These patients lose considerable water and energy through their skin…Children should be encouraged to eat and drink more than their unaffected sibs…I have a low threshold for recommending protein and calorie supplementation…Itching is usually mild; increased itching should prompt a search for chronic fungal infection. Most of my patients prefer emollients (Vaseline/aquaphor) to keratolytics/humectants. Early referral to the patient support group F.I.R.S.T. can be very helpful. LM
|
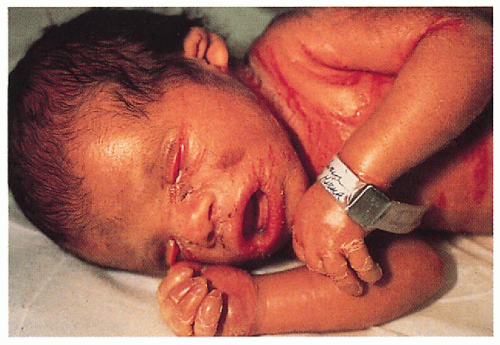
 1.9. Collodion baby at 2 weeks of age (1). |
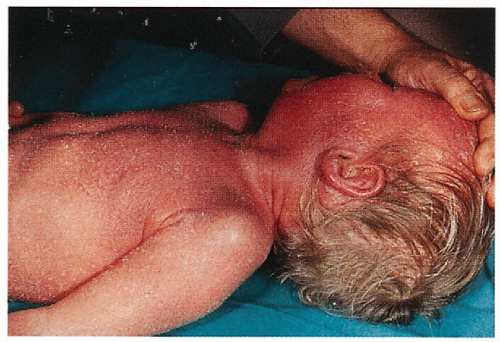 1.10. Erythroderma with fine, white scale on young boy (6). |
Harlequin Fetus
Inheritance
Autosomal recessive most likely; genetic heterogeneity with recent description of de novo deletion of 18q21
Prenatal Diagnosis
Amniocentesis—abnormal morphology of amniotic fluid cells
Ultrasound
Fetal skin biopsy—electron microscopy with absent lamellar bodies
Incidence
Less than 1:300,000; M=F
Age at Presentation
Birth
Pathogenesis
Heterogeneous molecular and genetic causes have been described; all patients have the following in common: defective keratinization with abnormal keratinocyte biochemical and morphologic differentiation and excessive hyperkeratosis; an error in lipid metabolism with lipid accumulation in stratum corneum; absent normal lamellar granules, defective profilaggrin conversion to filaggrin and a decrease in calpain (a calcium-activated protease important in calcium-mediated signaling and normal differentiation) may play a role in phenotype; 3 subtypes have been described based on different keratin protein expression, profilaggrin presence and size and number of lamellar granules
Key Features
Skin
Massive hyperkeratotic plates with deep fissures encasing newborn
Severe ectropion, eclabium, absent/deformed ears, nose, fingers, toes; poor temperature regulation
Generalized scaling with erythroderma in survivors of the neonatal period
Differential Diagnosis
Severe congenital ichthyosiform erythroderma (p. 12)
Severe lamellar ichthyosis (p. 10)
Laboratory Data
Sepsis workup
Management
Transfer to neonatal intensive care unit—monitor fluids, electrolytes, and for sepsis; systemic antibiotics, humidified incubators
Retinoids may help shed scale and contribute to survival;
If survival beyond neonate, referral to surgeon—correction of ectropion, hand/feet deformities; referral to dermatologist—-retinoids, emolliation
Referral to ophthalmologist—manage ectropion, secondary keratitis
Prognosis
If not stillborn, most die within the first few days of life as a result of sepsis or respiration and feeding complications from severe constriction of the chest and abdomen; survival has been reported with retinoid therapy
Clinical Pearls
Is not the uniformly fatal disease once thought…Support in a good pediatric intensive care unit (PICU) seems most critical for survival past postnatal period…Early intervention with oral retinoids may facilitate shedding of thick natal scale, but there are well-documented cases of survival without retinoids…Barrier is extremely poor so these kids need emollients 4-5 times a day…Call F.I.R.S.T. for names of physicians with experience. LM
|
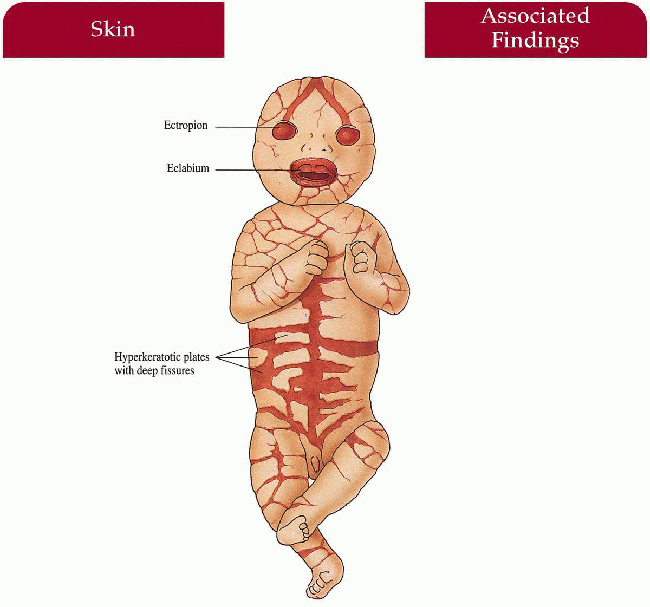
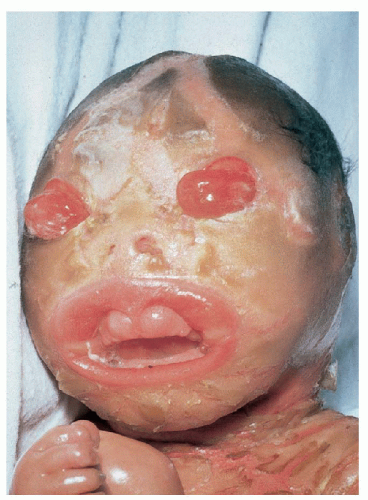 1.11. Newborn with severe eclabium, ectropion, deep fissures. (7) |
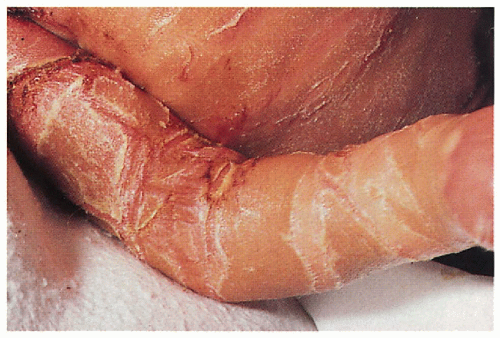 1.12. Close-up of thick, hyperkeratotic plates. (7) |
Sjögren-Larsson Syndrome
Inheritance
Autosomal recessive; Fatty aldehyde dehydrogenase (FALDH) gene 17p11.2
Prenatal Diagnosis
CVS/amniocentesis: fatty aldehyde dehydrogenase or fatty alcohol oxidoreductase assay; DNA mutation analysis if gene defect is known
Fetal skin biopsy at 23 weeks
Incidence
More than 200 cases reported, many from northern Sweden
Age at Presentation
Infancy (ichthyosis); by age 2-3 years old (central nervous system [CNS])
Pathogenesis
Over 50 mutations in the FALDH gene have been identified leading to a decrease in fatty-alcohol: NAD oxidoreductase (FAO) activity and subsequent defective conversion of fatty alcohol to fatty acid; this pathway is important in epidermal lipid synthesis as well as catabolism of phospholipids and sphingolipids in CNS myelin; accumulation of fatty alcohol, fatty aldehyde-modified lipids and leukotriene B4, which contributes to pruritus
Key Features
Skin
Infancy
Generalized ichthyosis with erythroderma, areas of fine scaling, areas of large lamellar scaling, hyperkeratosis, pruritus
After Infancy
Generalized darker scale without erythema accentuated in flexures, lower abdomen and back/sides of neck; spares central face
Central Nervous System
Mental retardation, spastic di-tetraplegia with scissor gait, speech deficits, epilepsy
Eyes
Atypical retinal pigment degeneration in macula—glistening white dots in a perimacular distribution; (many but not all cases) retinal pigmentary changes in some patients
Differential Diagnosis
Lamellar ichthyosis (p. 10)
Congenital ichthyosiform erythroderma (p. 12)
NLSD
Multiple sulfatase deficiency
Laboratory Data
Enzyme assay in cultured fibroblasts; DNA mutation analysis if defect known
Management
Referral to dermatologist—emolliation, retinoids
Referral to neurologist, ophthalmologist, orthopedist
Zileuton inhibits leukotriene B4 synthesis and may help pruritus
Prognosis
Dependent on severity of CNS complications—if wheelchair-bound and severely retarded, prognosis is guarded; otherwise patients typically live well into adulthood
Clinical Pearls
Pruritis in Sjögren-Larsson syndrome (SLS) is a distinguishing feature from other clinically similar diseases and it may respond to Zileuton therapy, which reduces leukotriene B4 levels…In some patients, the ichthyosis may wax and wane every few weeks…Photophobia is a common complaint…Retinal glistening white dots, if present in a patient with suspected SLS, is a reliable diagnostic sign, but its absence does not rule out SLS…Alopecia or nail dystrophy in not seen in this disease…The neurologic symptoms of SLS are not typically progressive, though spasticity symptoms may worsen with age if the patient does not get regular physical therapy…Anecdotal reports that the ichthyosis improves with administration of a low-fat diet supplemented with medium-chain fatty acids have not been reproduceable. WR
|
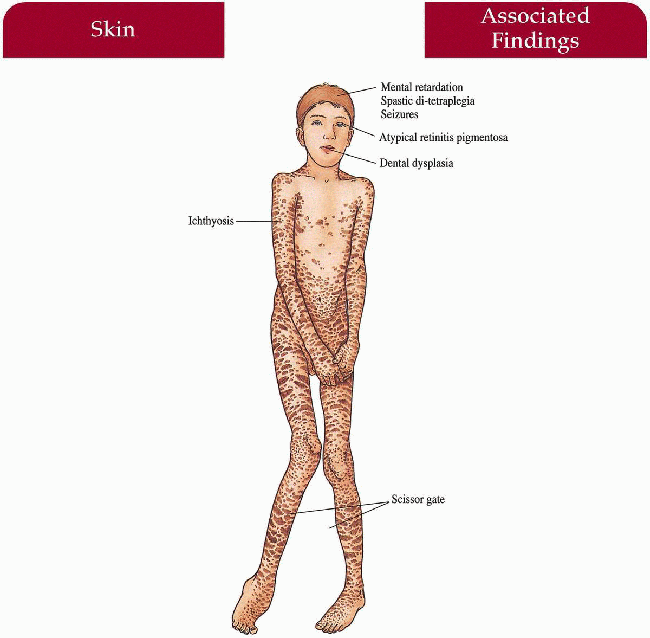
 1.13. Patients with ichthyosis, moderate spasticity and paresis (8). |
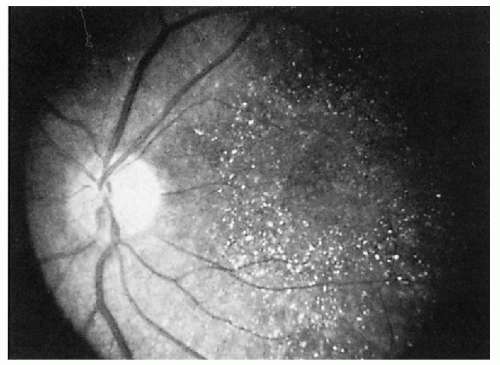 1.14. Atypical retinitis pigmentosa with “glistening dots” patterns (9). |
Refsum Syndrome
Synonym
Phytanic acid storage disease
Heredopathia atactica polyneuritiformis
Inheritance
Autosomal recessive; PAHX gene on 10p, PEX7 gene on 6q
Prenatal Diagnosis
CVS/amniocentesis: phytanic acid oxidase assay on cultured cells; DNA analysis
Incidence
Rare; approximately 100 cases reported; M=F
Age at Presentation
Neurologic symptoms start in childhood; cutaneous changes usually occur as an adult
Pathogenesis
Mutations in the PAHX gene create a deficiency in phytanoyl-CoA hydroxylase, a peroxisomal enzyme responsible for the catalyzation of phytanic acid; deficient enzyme leads to an accumulation of phytanic acid in serum and replacement of the normal fatty acids in epidermal lipids and other tissues throughout the body; can also be caused by mutations in the PEX7 gene that encodes peroxin 7, a receptor important in targeting enzymes to peroxisomes; defective PEX7 leads to a deficiency in multiple peroxisomal enzymes
Key Features
Skin
Mild ichthyosis (i.e., ichthyosis vulgaris) usually beginning after neurologic symptomatology
Central Nervous System
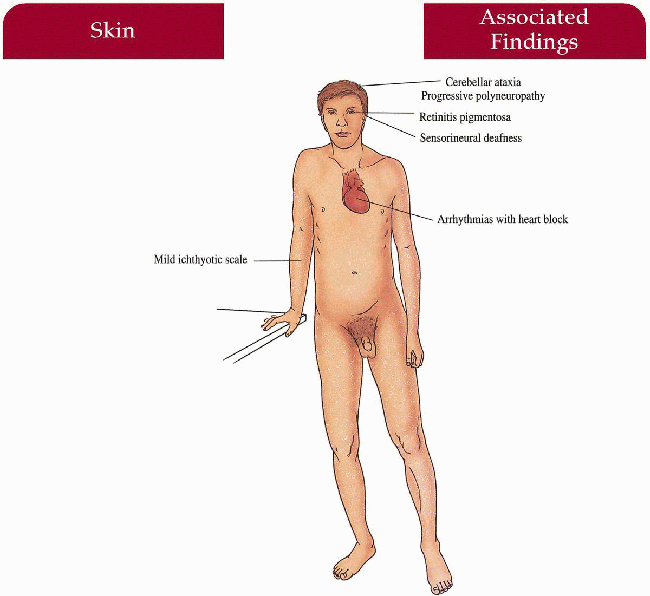
Cerebellar ataxia, progressive peripheral polyneuropathy
Eyes
Retinitis pigmentosa with salt and pepper pigment, secondary night blindness
Ear-Nose-Throat
Sensorineural deafness
Cardiac
Arrhythmias with heart block, cardiac failure
Musculoskeletal
Symmetric muscular wasting, variety of skeletal anomalies
Differential Diagnosis
Peroxisomal deficiency disorders
Ichthyosis vulgaris (p. 2)
Vitamin B deficiency
Laboratory Data
Increased serum phytanic acid; decreased phytanic acid oxidase activity in cultured fibroblasts
Skin biopsy revealing lipid-filled vacuoles in basal keratinocytes
Increased cerebrospinal fluid (CSF) protein without cells
Management
Dietary restriction of phytanic acid-decrease green vegetables, dairy products and ruminant fats
Plasma exchange removal of phytanic acid
Referral to neurologist, ophthalmologist, cardiologist, dermatologist, otolaryngologist, and physiatrist
Prognosis
If diet and exchange instituted early on, progression of disease can be halted; if untreated, symptomatology is progressive with remissions and exacerbations culminating in premature sudden death from cardiac arrythmias (heart block) or respiratory failure (medullary depression)
Clinical Pearls
Signs and symptoms are very dependent on diet; therefore onset and severity are very variable…Neurologists usually make the diagnosis…Ichthyosis is usually mild and responsive to diet…These low vegetable/animal fat diets are tough to follow. LM
|
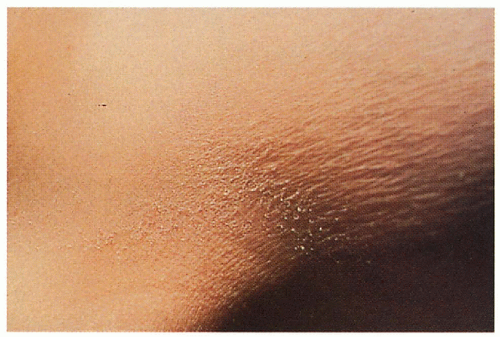 1.15. Fine, white scales in flexures (2). |
 1.16. Retinitis pigmentosa with typical “salt-andpepper” pattern (10). |
Conradi-Hünermann Syndrome
Synonym
X-linked dominant chondrodysplasia punctata; Conradi-Hunermann-Happle syndrome
Inheritance
X-linked dominant; Emopamil-binding protein (EBP) gene on Xp11
Prenatal Diagnosis
Ultrasound evaluation of long bones
Incidence
Rare; usually lethal in males; reports of surviving males both with/without 47, XXY
Age at Presentation
Birth
Pathogenesis
Mutation in the EBP gene or 3β-hydroxysteroid-Δ8-Δ7-isomerase leads to a defect in cholesterol biosynthesis and can explain skeletal phenotype
Key Features
Skin
Ichthyosiform erythroderma in Blaschko’s lines in infancy; resolves with follicular atrophoderma and/or hyperpigmentation
Hair
Coarse, patchy alopecia
Eyes
Asymmetric focal cataracts
Musculoskeletal
Stippled epiphyses (punctate calcifications), asymmetric limb shortening, short stature, scoliosis
Cranofacial
Frontal bossing, macrocephaly, flat nasal root, asymmetric
Central Nervous System
Mental retardation (rare)
Differential Diagnosis
Autosomal recessive rhizomelic chondrodysplasia punctata
X-linked recessive chondrodysplasia punctata with steroid sulfatase deficiency
CHILD syndrome
Incontinentia pigmenti
Laboratory Data
Bone films
Neonatal skin biopsy—may reveal calcium in the epidermis with von Kossa’s stain
Peroxisomal function in cultured fibroblasts
Management
Referral to orthopedist, dermatologist, ophthalmologist
Examine first-degree relatives
Prognosis
Ichthyosis and stippled epiphyses resolve after infancy; orthopedic complications predominate with normal life span
Clinical Pearls
If an erythrodermic baby has hyperkeratosis in a linear or whorled pattern, look for epiphyseal stippling on x-ray…By several years of age, the epiphyseal stippling is usually gone and the erythema more limited…Severity of skin and skeletal disease varies greatly between individual adults, and there are no good early predictors yet…Follicular atrophoderma seems to be a nearly constant finding in adults. LM
|
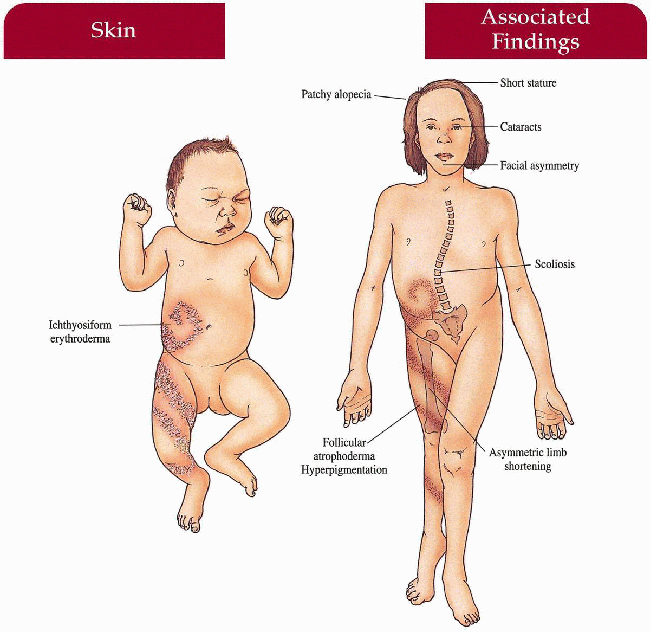
 1.17. Infant with ichthyosiform erythroderma in Blaschko’s lines (11). |
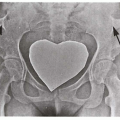
 1.18. Stippled epiphyses (12). |
CHILD Syndrome
Synonym
Congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD)
Unilateral congenital ichthyosiform erythroderma
Inheritance
X-linked dominant; NSDHL gene on Xq28
Prenatal Diagnosis
Ultrasound detection of limb/organ defects
Incidence
Rare; lethal in males
Age at Presentation
Birth to 1 month old
Pathogenesis
Mutations in the NSDHL gene encoding for 3β-hydroxysteroid dehydrogenase are most common; EBP gene defect (i.e. Conradi’s) has been described; both enzymes involved in cholesterol biosynthesis
Key Features
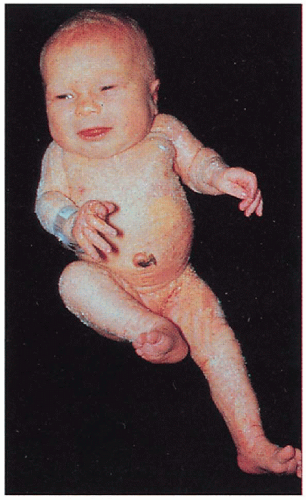
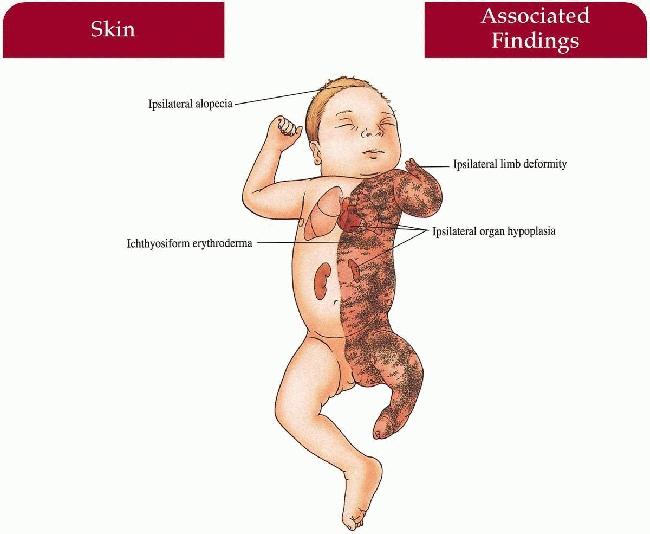
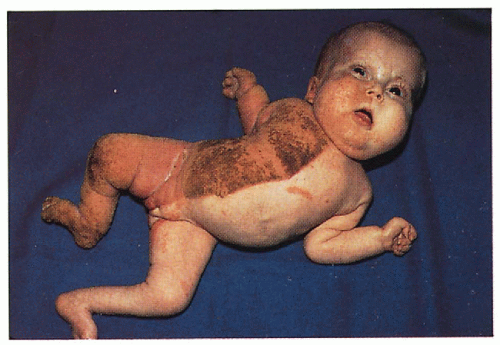
Skin
Unilateral ichthyosiform erythroderma with sharp midline cutoff involving trunk and limbs; ± linear or segmental involvement on contralateral side; lesions tend to improve with age but may persist in skin folds (ptychotropism)
Hair
Ipsilateral alopecia
Nails
Severe dystrophy
Musculoskeletal
Hypoplasia to agenesis of limbs ipsilateral to ichthyosis; other ipsilateral bones may be involved; ± stippled epiphyses
Internal Organs
Hypoplasia to agenesis of organs below ichthyosis-variety of organs reported including CNS, cardiovascular, renal, and genitourinary involvement
Differential Diagnosis
Conradi-Hünermann syndrome
Inflammatory linear verrucous epidermal nevus
Laboratory Data
Computed tomography/magnetic resonance imaging (CT/MRI) scan of ipsilateral side
Management
Referral to dermatologist—topical therapy
Referral to orthopedist
Referral to organ-specific subspecialist
Prognosis
Dependent on which organs are affected—can range from normal life span to incompatible with life
Clinical Pearls
Experienced clinicians made a connection long ago between the constellation of findings in CHILD and Conradi-Hünermann, so it was satisfying to learn that their gene defects share a common biochemical pathway…In CHILD syndrome, the skin lesions can look like inflammatory linear epidermal nevi…Excision can be curative for individuals with relatively narrow, nevoid bands. LM
|
 1.19. Unilateral ichthyosiform erythroderma with ipsilateral limb hypoplasia (13). |
 1.20. Close-up of thick hyperkeratosis of the left foot and leg (13). |
Netherton Syndrome
Synonym
Ichthyosis linearis circumflexa (ILC)
Inheritance
Autosomal recessive; SPINK5 gene on 5q32
Prenatal Diagnosis
DNA mutation analysis if defect known in family
Incidence
Rare with a few dozen case reports; M=F; observed M:F=1:2
Age at Presentation
Birth to first few months of life
Pathogenesis
Mutations in SPINK5 gene encoding LEKT1, a serine protease inhibitor that may be important in downregulating inflammatory pathways; LEKT1 also associated with atopy
Key Features
Skin
Birth-Few Months
Generalized erythema and scaling with secondary hypernatremia, failure to thrive
Later in Infancy
Migratory erythematous, polycyclic, serpiginous plaques with double-edged scale along the margins (ILC)
Atopic dermatitis with flexural lichenification and pruritus
Seborrheic-like scale and erythema on face, scalp, eyebrows
Hair
Trichorrhexis invaginata (ball-and-socket configuration; bamboo hair)—most characteristic; may also have pili torti or trichorrhexis nodosa; eyebrow hair may be most common site
Short, sparse
Immunology
Anaphylactic reactions to foods
Differential Diagnosis
Congenital ichthyosiform erythroderma (p. 12)
Seborrheic dermatitis
Dermatophytosis
Laboratory Data
Light microscope—examination of hair shaft
Increased serum immunoglobulin E (lgE)
KOH
Management
Monitor in infancy for hypernatremia, and failure to thrive
Referral to dermatologist—topical therapy, retinoids; avoid keratolytics (can worsen condition) and tacrolimus ointment (increased absorption from compromised skin barrier with increased risk of toxicities)
Referral to allergist—radioallergosorbent assay test (RAST)
Prognosis
May have partial remissions and may improve at puberty; normal life span
Clinical Pearls
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


