Disorders of Connective Tissue
Juoni Uitto M.D., Ph.D.
Ilona Frieden M.D.
Kurt Hirschhorn M.D.
Judith Willner M.D.
Clinical Pearls
(JU)
(IF)
(KH)
(JW)
Ehlers-Danlos Syndrome
Inheritance
Type
Classical (I and II):
autosomal dominant; gene locus 2q31, 9q34
Hypermobility (III):
autosomal dominant
Vascular (IV):
autosomal dominant; gene locus 2q31
Kyphoscoliosis (VI):
autosomal recessive; gene locus 1p36.3-p36.2
Arthrochalasia:
autosomal dominant (VIIA, VIIB); gene locus 7q22.1, 17q21-22
Dermatosparaxis (VIIC):
autosomal recessive ; gene locus 5q23
Other variants
(V, VIII, X, XI)
Prenatal Diagnosis
Classical
Chorionic villus sampling (CVS)/amniocentesis-—deficient type V collagen in cultured cells
DNA mutation analysis
Vascular
CVS/amniocentesis—decreased type III collagen in cultured cells
DNA analysis
Kyphoscoliosis
Amniocentesis—decreased lysyl hydroxylase activity in cultured amniocytes
DNA analysis
Arthrochalasia/Dermatosparaxis
CVS/amniocentesis—deficient type I collagen in cultured cells
Incidence
Approximately 1:5,000; M=F, except X-linked (all male)
Classical
Approximately 80% of all Ehlers-Danlos syndrome (EDS)
Hypermobility
Approximately 10% of all EDS
Vascular
Approximately 4% of all EDS
All other types comprise remaining 6%
Age at Presentation
Birth to early childhood
Pathogenesis
Classical
Mutations in COL5A1 and COL5A2 chains in type V collagen account for about 50% of cases; deficiency in tenascin X in 3% of patients
Vascular
Mutations in COL3A1 results in abnormal synthesis, structure and secretion of type III collagen
Kyphoscoliosis
Mutation in procollagen lysyl 2-oxoglutarate 5 dioxygenase (PLOD) gene leads to deficient lysyl hydroxylase
Arthrochalasia
Mutations involving the amino terminal propeptide cleavage sites of COL1A1 (type A) or COL1A2 (type B) leads to defective conversion of procollagen to collagen type I
Dermatosparaxis
Recessive mutations in the type I collagen N-peptidase gene
Hypermobility
Unknown
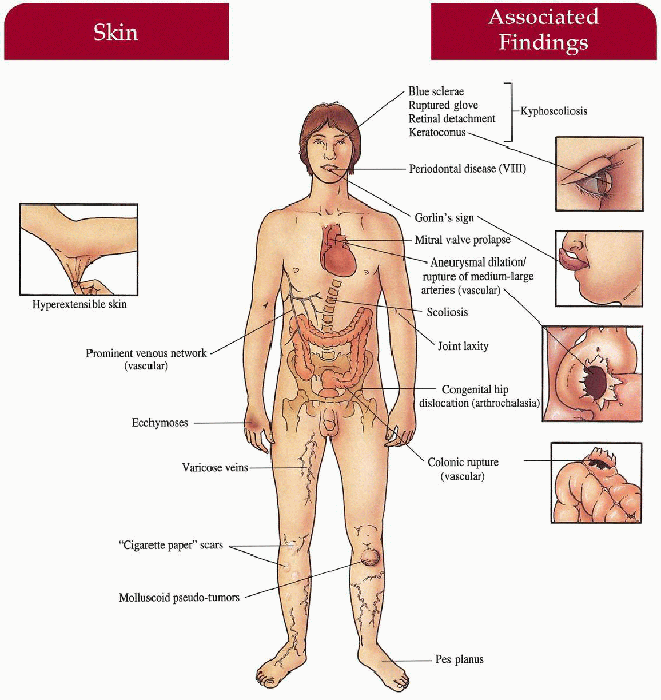
Key Features
Classical (I, II)
Skin
Hyperextensible with “snap-back” elasticity, gaping wounds from minimal trauma, “cigarette-paper” scars, molluscoid pseudotumors, calcified subcutaneous nodules, varicose veins, ecchymoses
Musculoskeletal
Hypermobile joints with potential delay in ambulation, recurrent joint dislocations, pes planus, genu recurvatum, kyphoscoliosis, inguinal/umbilical hernias
Cardiovascular
Mitral valve prolapse
Craniofacial
Epicanthic folds, hypertelorism, blue sclerae, + Gorlin’s sign (ability to touch nose with tongue tip)
Pregnancy
Prematurity caused by early rupture of fetal membranes in affected fetus; postpartum hemorrhage
Hypermobility (III)
Musculoskeletal
Severe joint laxity with delay in ambulation, recurrent dislocations, early-onset degenerative joint disease
Skin
Minimally affected with mild hyperextensiblity
Cardiovascular
Mitral valve prolapse
Vascular (IV)
Skin
Thin, translucent, fragile with easily seen venous network; inextensible, ecchymoses, varicose veins
Musculoskeletal
Minimal joint laxity (hands and feet)
Arterial
Aneurysm, dissection, rupture of large and medium-sized vessels; arteriovenous fistulas
Gastrointestinal
Colonic rupture, recurrent abdominal pain
Pregnancy
Uterine rupture, arterial rupture, tearing of vaginal tissues
Craniofacial
Acrogeric facies
Kyphoscoliosis (VI)
Skin
Hyperextensible, fragile, ecchymoses
Musculoskeletal
Newborn hypotonia, joint laxity, severe kyphoscoliosis
Eyes
Ruptured globe, retinal detachment, intraocular hemorrhage, keratoconus, blindness
Arthrochalasia (VIIA, B)
Skin
Mild hyperextensibility, fragility
Musculoskeletal
Congenital hip dislocation, severe joint hypermobility with dislocation of large and small joints, scoliosis, short stature
Dermatosparaxis (VIIC)
Skin
Severe fragility, laxity with sagging redundancy; easy bruisability, umbilical/inguinal hernias, premature rupture of fetal membranes; normal wound healing
Other Variants
Type VIII
Skin
Hyperextensible, fragile, ecchymoses; pretibial, yellow-brown, wrinkled scarring
Mouth
Severe periodontitis with resorption of alveolar bone and premature loss of permanent teeth
Type X
Skin
Mild hyperextensibility, petechiae, ecchymoses
Musculoskeletal
Joint laxity
Type XI
Musculoskeletal
Large joint (especially hips, shoulders, and patella) laxity with recurrent dislocations
Laboratory Data
Vascular
Skin biopsy—biochemical assay revealing decreased type III collagen in cultured fibroblasts
Two-dimensional echocardiography
Kyphoscoliosis
Skin biopsy: biochemical assay revealing reduced lysyl hydroxylase activity in cultured fibroblasts
Arthrochalasia/Dermatosparaxis
Electrophoresis reveals procollagen alpha1(I) or alpha 2(I) chains from cultured fibroblasts or collagen
Management
General
Referral to dermatologist, orthopedic surgeon
Skin protection from trauma
Advise surgeons regarding poor wound healing
Advise obstetrician regarding prematurity
Examine first-degree family members
Vascular
Referral to cardiologist/cardiovascular surgeon
Advise obstetrician regarding pregnancy avoidance/potential labor complications
Avoid arteriography
Avoid physical contact sports
Referral to gastroenterologist if symptomatic
Kyphoscoliosis
Referral to ophthalmologist
Oral ascorbic acid
Type VIII
Referral to dentist
Type X
Referral to hematologist
Prognosis
Normal life span except in vascular type with potential for premature death in second to third decade because of complications from arterial or colonic rupture or maternal death because of uterine or arterial rupture; kyphoscoliosis type may cause blindness
Clinical Pearls
This syndrome continues to be a challenge to practicing dermatologists. The wide spectrum of phenotypic manifestations blend at one end to the constitutive features in the general population and at the other end can be a cause of early demise. The molecular basis of all six major forms of EDS has now been deciphered, the mutations residing either in the genes encoding collagens, enzymes modifying the primary collagen translation products, or tenascin-X, another connective tissue protein.
The major type of EDS not to be missed is the vascular type (old EDS IV). They are at high risk for arterial and intestinal ruptures, and often unexpectedly, to uterine rupture during labor, with catastrophic consequences. As soon as you establish this diagnosis, get a cardiovascular evaluation, paying attention to the aortic root and possible aneurysms.
Another type not to miss is the kyphoscoliosis type (old EDS VI). In some cases one may be able to overcome the enzyme deficiency simply by supplementary ascorbic acid in the diet. In fact, some physicians advocate ascorbic acid for all EDS types.
The previous EDS IX has been excluded from EDS category and is known as the occipital horn syndrome (because of the characteristic bone protrusions at the base of the skull). It is allelic with Menkes syndrome.
General treatment of EDS is protection from trauma, use of shin guards when playing sports, avoid sharp edges of furniture at home. Loose jointedness, particularly affecting knees and hips, can result in early osteoarthrosis; a pediatric orthopedic consultation may be helpful. Mitral valve prolapse is exceedingly common among patients with EDS. JU
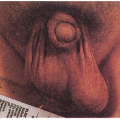
 4.1. Hyperextensible skin. (55) |
 |
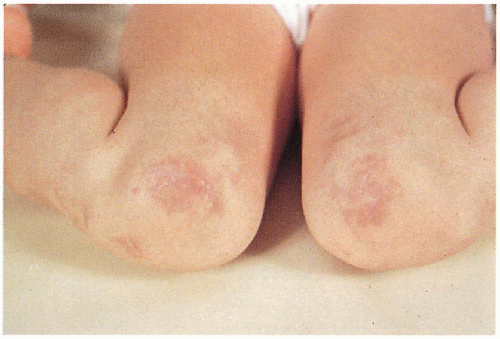 4.3. “Cigarette paper” atypical scars on knees. (55) |
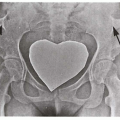
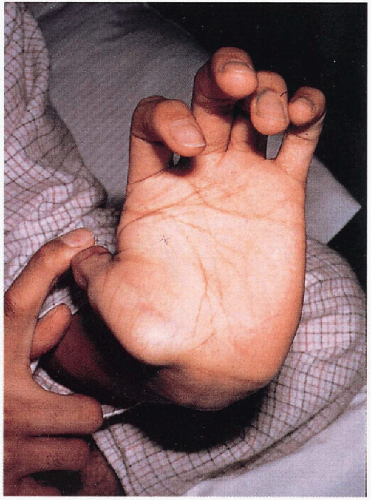 4.4. Hypermobile joints. (5) |
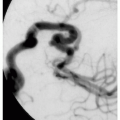
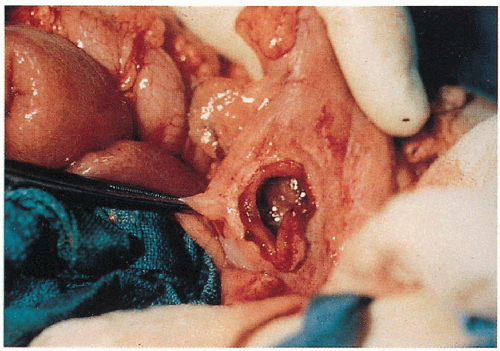 4.5. Sigmoid perforation in a patient with vascular EDS. (56) |
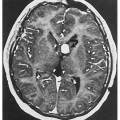
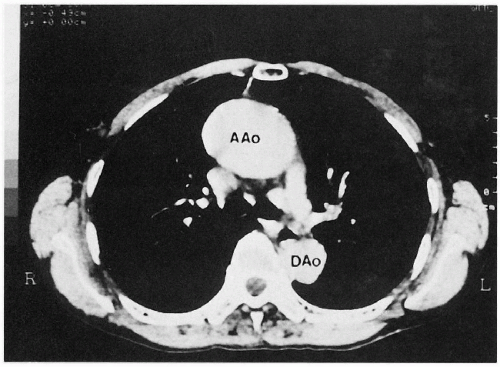 4.6. CT with contrast reveals marked dilation of the ascending aorta in a patient with vascular EDS. (57) |
|
Marfan Syndrome
Inheritance
Autosomal dominant; mutation in fibrillin-1 on chromosome 15
Prenatal Diagnosis
DNA analysis
Incidence
1:10,000-20,000; M=F
Age at Presentation
Infancy if suspected by family history; usually second or third decade of life
Pathogenesis
Mutation in fibrillin gene, coding for a vital component of the microfibrillar system, results in a lack of fibrillin with concomitant defects in the ocular, cardiovascular, and musculoskeletal system
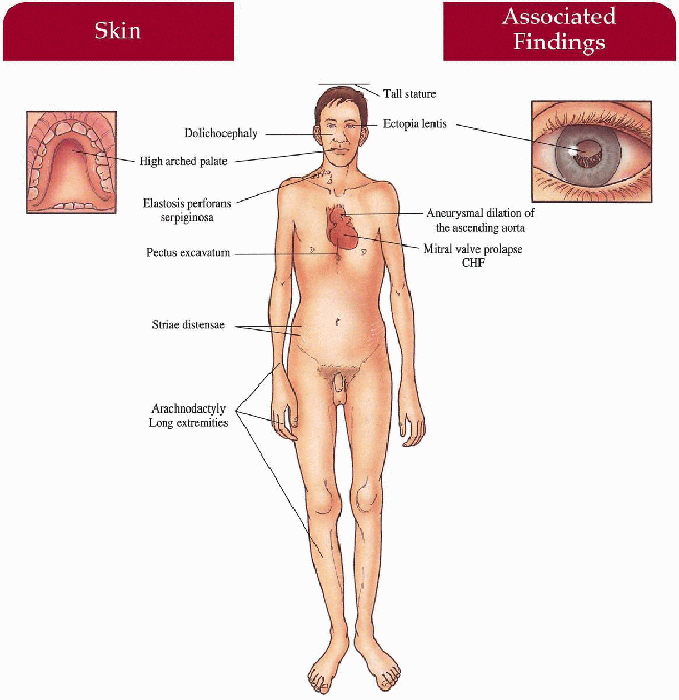
Key Features
Musculoskeletal
Tall stature, lower body length longer than upper body length, arachnodactyly, dolichocephaly, pectus excavatum, high-arched palate, loose joints, poor muscle tone, kyphoscoliosis, pes planus, inguinal hernia
Eyes
Ectopia lentis (upward displacement in 75%)
Myopia
Cardiovascular
Progressive aneurysmal dilatation of ascending aorta with secondary regurgitation, congestive heart failure (CHF), dissection and rupture
Mitral valve prolapse
Skin (less common)
Striae distensae
Elastosis perforans serpiginosa
Decreased subcutaneous fat
Differential Diagnosis
Congenital contractural arachnodactyly
Multiple endocrine neoplasia type IIb (p. 190)
Homocystinuria (p. 332)
Stickler syndrome
Laboratory Data
Echocardiagram
Chest x-ray
Management
Referral to cardiologist/cardiac surgeon—surgical repair, β-blockers
Referral to ophthalmologist
Referral to orthopedic surgeon
Estrogen therapy—prevent excessive tallness in females
Prognosis
Although the prognosis has improved dramatically with advanced cardiovascular surgical repair, patients may die prematurely from cardiac complications; marked variability in severity
Clinical Pearls
This systemic connective tissue disorder is caused by mutations in the fibrillin-1 gene. The phenotypic variability reflects the types of mutations and their consequences at the mRNA and protein levels. The major complications relate to cardiovascular findings, including aortic dilatation, dissection and aneurysms. The patients should be followed closely by cardiovascular surgeons. Lowering blood pressure and using beta-blockers is clearly helpful. The eye problems require regular follow-up by an ophthalmologist. JU
|
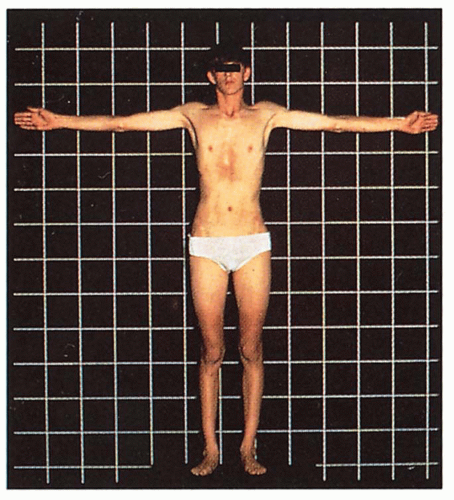 4.7. Tall stature with long extremities and pectus excavatum. (48) |
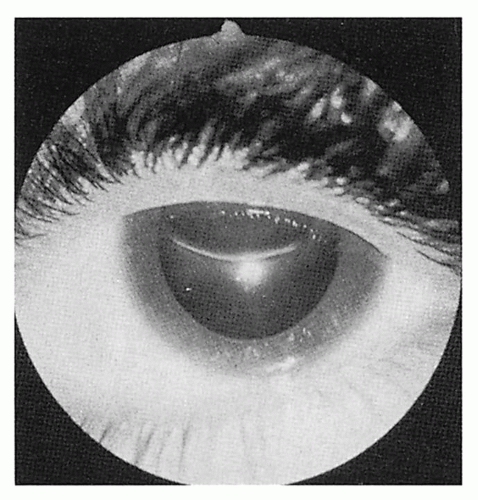 4.8. Upward displacement of lens. (58) |
Cutis Laxa
Synonym
Generalized elastolysis
Inheritance
Autosomal recessive (most common)-FBLN5 (fibulin 5) gene on 14q32; another locus on 5q23-31; autosomal dominant—elastin gene on 7q11 and FBLN5 on 14q32 (usually skin only); X-linked recessive—ATP7A on Xq12-13; acquired
Prenatal Diagnosis
DNA mutation analysis
Incidence
Rare
Age at Presentation
Birth to infancy
Pathogenesis
Heterogeneous mutations in fibulin 5 gene, elastin gene, or ATP7A gene (adenosine triphosphatase [ATPase] mutation that impairs copper transport necessary for lysyl oxidase activity and normal elastin production) contributes to variability in clinical severity
Key Features
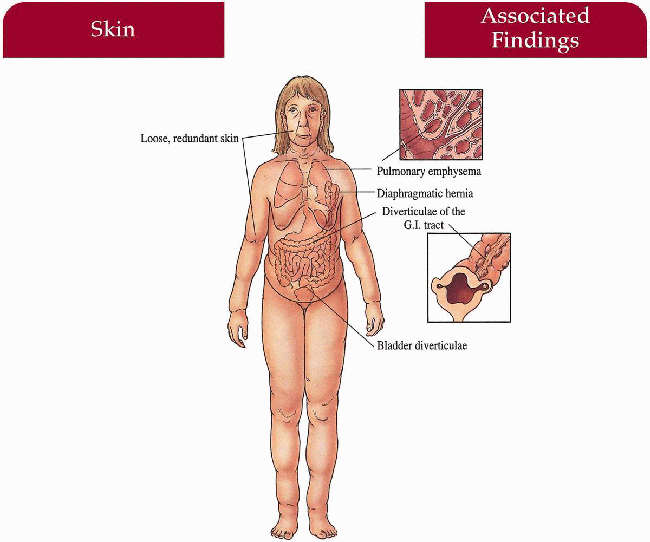
Skin
Loose, redundant, pendulous skin folds with hound-dog facies, often generalized; inelastic, lacks recoil
Premature aged appearance
Oral
Vocal cord laxity causing deep, resonant voice
Lungs
Newborn—hypoplastic lungs
Emphysema (autosomal recessive)—may be complicated by tachypnea, pneumonitis, cor pulmonale
Gastrointestinal (autosomal recessive)
Esophageal, duodenal, rectal diverticulae
Genitourinary (autosomal recessive and x-linked)
Bladder diverticulae
Musculoskeletal (autosomal recessive and x-linked)
Inguinal, diaphragmatic, umbilical hernia, hip dislocation, occipital horn exostoses (x-linked)
Differential Diagnosis
Pseudoxanthoma elasticum (p. 144)
EDS (p. 134)
Granulomatous slack skin
DeBarsy syndrome
SCARF syndrome
Laboratory Data
Skin biopsy—decreased, fragmented elastic fibers visualized with Verhoeff-van Gieson stain
Serum copper and ceruloplasmin levels
Chest x-ray
Management
Referral to plastic surgeon
Referral to pulmonologist, gastroenterologist, urologist, surgeon if symptomatic
Sunscreen protection
Prognosis
Great variability in severity ranging from death in neonate if born with hypoplastic lungs to only skin involvement with normal life span if no pulmonary disease (majority with latter).
Clinical Pearls
The underlying pathology relates to perturbation in the elastic fiber network, and mutations have been demonstrated both in the elastin and fibulin-5 genes. The clinical spectrum spans from relatively mild skin involvement to extremely severe with skin problem associated with extracutaneous manifestations, including pulmonary emphysema, arterial aneurysms, and vesico-urinary diverticula. Therefore, careful systemic evaluation, especially in patients with congenital forms of cutis laxa, is in order. The inheritance can be either autosomal dominant or autosomal recessive. Acquired, late-onset cutis laxa is often associated with urticarial and/or inflammatory lesions and is sometimes a sequela of acute drug reaction, such as to penicillin. In the latter case, inflammatory cells that contain powerful elastases degrade elastin in the skin, resulting in paucity of elastic fibers. As a result, the entire skin shows progressive sagging, and because of relatively poor repair capacity, new elastic fibers are not formed in adult skin and the sagging is permanent. Solar elastosis tends to aggravate this condition, and sunscreen application should be stressed. Facelifts do provide improvement, but this may be only temporary. JU
|
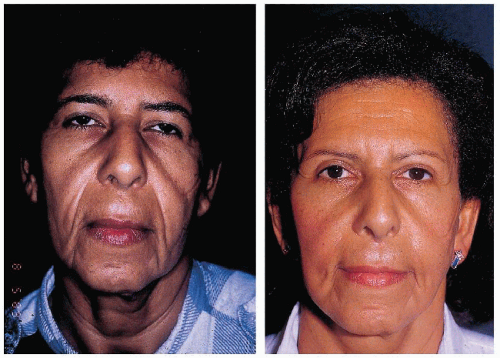 4.9. Left: Twenty-three years-old patient with loose, pendulous skin folds. Right: Same patient at 38 y.o. after 3 face-lifts, earlobe correction, and resection of excess skin from the nasolabial folds and upper lip. (59) |
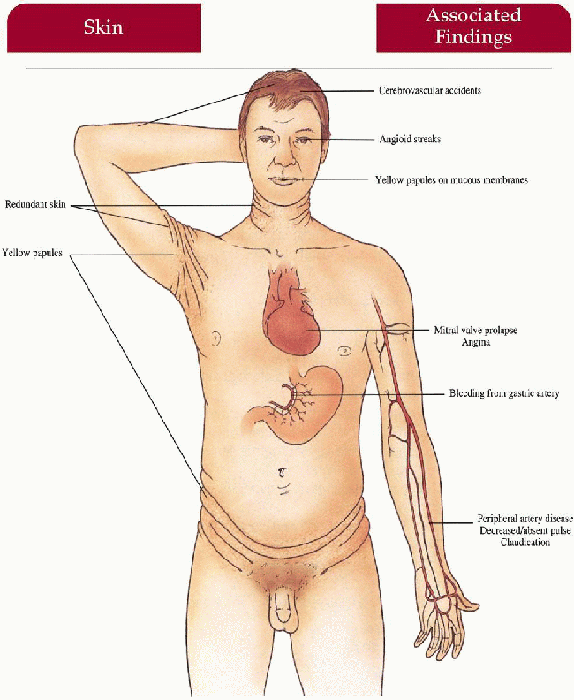
Pseudoxanthoma Elasticum
Inheritance
Autosomal recessive (most common); autosomal dominant; ABCC6 (adenosine triphosphate [ATP]-binding cassette subfamily C member 6) transporter gene on 16p13
Prenatal Diagnosis
DNA mutation analysis if defect known
Incidence
Approximately 1:100,000; M=F
Age at Presentation
Childhood to second or third decade of life
Pathogenesis
Mutation in ABCC6 transmembrane transporter gene that encodes a multidrug resistance protein; the correlation of this gene defect with the phenotype has not been elucidated
Key Features
Skin
Yellow papules coalescing to plaques overlying redundant, lax, soft skin folds on sides of neck, axillae, antecubital fossae, abdomen, groin, thighs
Mucous Membranes
Yellow papules on labial mucosa, soft palate, rectal and vaginal mucosa
Eyes
Angioid streaks (rupture in Bruch’s membrane secondary to elastic fiber defect), macular degeneration, retinal hemorrhage causing blindness, retinal pigmentation alteration
Cardiovascular
Gastric artery hemorrhage (common) with epistaxis, hematemesis; claudication, decreased/absent peripheral pulses, hypertension, angina pectoris, myocardial infarction, cerebrovascular accidents, mitral valve prolapse
Obstetrics
Increased first trimester miscarraige, increased cardiovascular complications
Differential Diagnosis
Cutis laxa (p. 142)
Angioid streaks: Sickle cell anemia, Paget’s disease of bone, hyperphosphatemia
EDS (p. 134)
Laboratory Data
Skin biopsy (affected, normal, or cicatricial skin)—Von Kossa stain (calcium), Verhoeff-van Gieson stain (curled elastin fibers)
Fundoscopy
X-ray of extremity or abdomen if symptomatic
Management
Complete physical examination and regular follow-up with primary care physician
Referral to dermatologist, ophthalmologist
Referral to gastroenterologist, cardiologist, neurologist if symptomatic
Referral to plastic surgeon—cosmetic correction
Advise obstetrician during pregnancy
Restriction of calcium intake (controversial)
Examination of first degree family members by dermatologist, ophthalmologist, cardiologist
Prognosis
Shortened life span secondary to cardiovascular complications
Clinical Pearls
This clinical entity can occasionally be a diagnostic problem, particularly because of delayed onset and considerable intrafamilial and interfamilial heterogeneity. Diagnosis can usually be confirmed by skin pathology, but recent identification of the mutated gene, ABCC6, provides a molecular tool to confirm the diagnosis. In cases without definitive cutaneous findings, but with angioid streaks and family history of pseudoxanthoma elasticum (PXE), mutation analysis can be used for presymptomatic diagnosis.
Patients need to be followed by ophthalmologists who may consider laser treatment of retinal hemorrhages. Avoidance of head trauma is important to prevent retinal bleeding. Although total blindness is extremely rare, some patients may become legally blind at a relatively early age. Plastic surgery may be helpful in improving the cosmetic appearance of skin. The major life-threatening problems are myocardial infarct and occasionally, massive gastrointestinal hemorrhages, particularly in families with predominant cardiovascular manifestations. Strongly advise patients against smoking, because it exacerbates intermittent claudication and other cardiovascular problems. Calcium intake is a controversial subject, as it has been suggested that high calcium intake during the childhood or early adolescence results in more severe phenotype. I do not recommend low-calcium diet for adults as it may accelerate development of osteoporosis, and balanced diet including modest amounts of dairy products should be fine for children as well.
Direct patients to the website of PXE International, a patient advocacy organization: www.pxe.org. JU
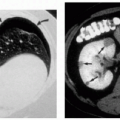
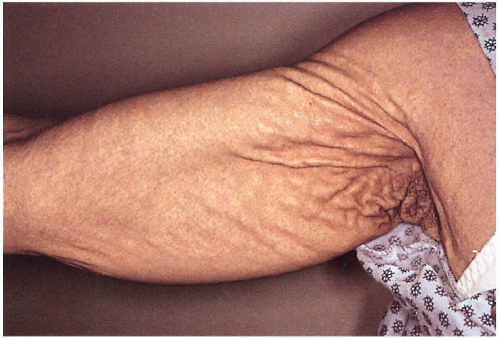 4.10. Axilla with coalescing yellow papules over redundant skin. |
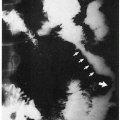
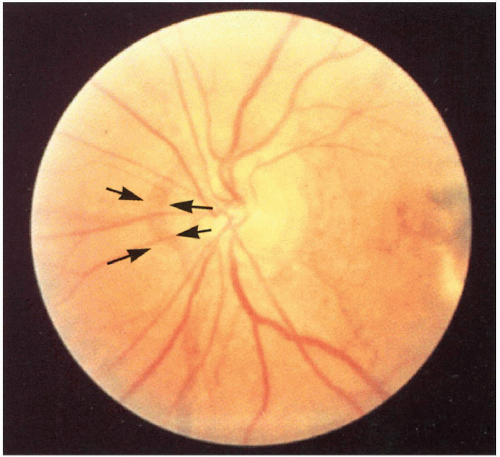 4.11. Angioid streaks (arrows). |
|
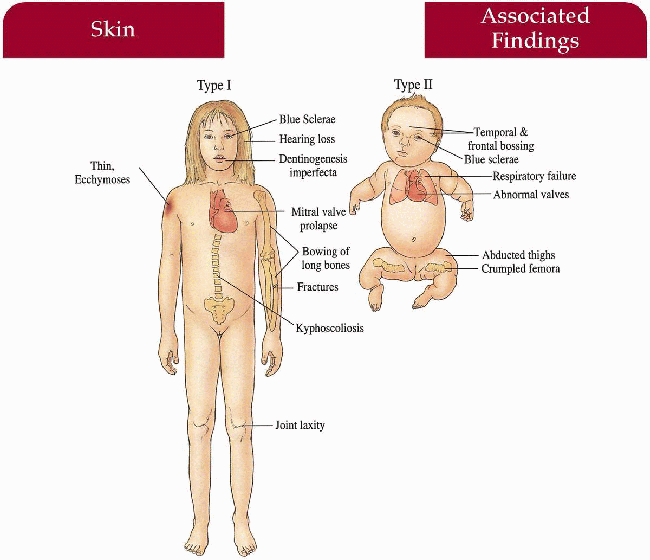
Osteogenesis Imperfecta
Inheritance
Type I—autosomal dominant
Type II—autosomal dominant and recessive
Type III—autosomal dominant and recessive
Type IV—autosomal dominant
COL1A2 gene on 7q22 and COL1A1 gene on 17q22
Prenatal Diagnosis
Ultrasonography/in utero x-ray at 16 weeks
DNA analysis
Incidence
1:5-10,000—type I most common; M=F
Age at Presentation
Birth to adulthood depending on type
Pathogenesis
Heterogeneous genetic mutations in genes encoding type I collagen (α1 and α2 chains) provides for heterogeneous phenotype; mutation may alter amount of collagen produced (milder phenotype) or change the structure of type I collagen (mild-severe phenotype)
Key Features
Skin
Thin, decreased elasticity; easy bruising (I, IV)
Eyes
Blue sclerae (I, II, III [infant])
Ear-Nose-Throat
Hearing loss secondary to otosclerosis
Musculoskeletal
I (mild/moderate)—fractures, bowing of long bones, joints lax, kyphosis
II (severe)—multiple fractures in utero; newborn with beaded ribs, crumpled humeri and femora; frontal, temporal bossing; limb avulsion during delivery, abducted thighs
III—fractures in utero, at birth; progressive kyphoscoliosis, bowing with crippling deformities
IV—fractures at birth and childhood with decreased frequency with age
Teeth
Dentinogenesis imperfecta (I, IV)
Cardiac
Mitral valve prolapse (I), aortic valve disease (I), autopsy reveals valvular disease (II)
Differential Diagnosis
Child abuse
Achondroplasia
Laboratory Data
Bone films
Echocardiography
Audiology examination
Management
Referral to orthopedist, psychiatrist, cardiologist, otolaryngologist, dentist if symptomatic
Intranasal calcitonin may reduce incidence of fractures
Bisphosphonate treatment
Prognosis
Variable with death in perinatal period (type II), increased mortality in third to fourth decade as a result of cardiorespiratory failure (type III), limb deformities after fractures, otherwise potential normal life span with limited morbidity
Clinical Pearls
Different mutations in type I collagen gene are responsible for varying severity of disease … Milder forms are often misdiagnosed as child abuse … However, osteogenesis imperfecta (OI) kids have thinner bones and fracture straight through, abused kids get spiral fractures … Blue sclerae should be looked for when evaluating for abuse … Can diagnose in utero with ultrasound … Looks like severe chondrodystrophies except you often see fractures … May need dentures as children … If teeth involved you usually have worse fractures … One of many dominant syndromes where you get an association with increased paternal age and a new mutation … Confirmation of prenatal and clinical diagnosis is available by collagen examination in cultured fibroblasts and by mutation screening. KH, JW
|
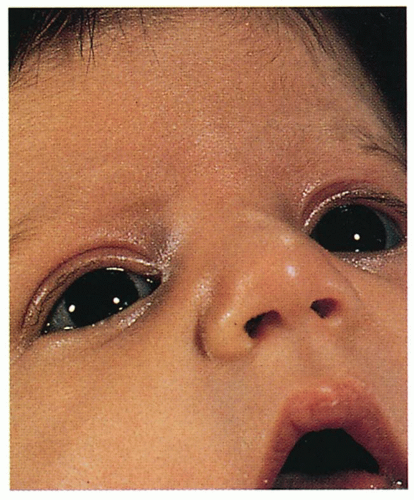 4.12. Blue sclerae. (60) |
 4.13. Severe bowing, kyphoscoliosis with crippling deformities. (60)
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|