Fig. 10.1
Poikilodermic cGVHD on the neck and upper chest
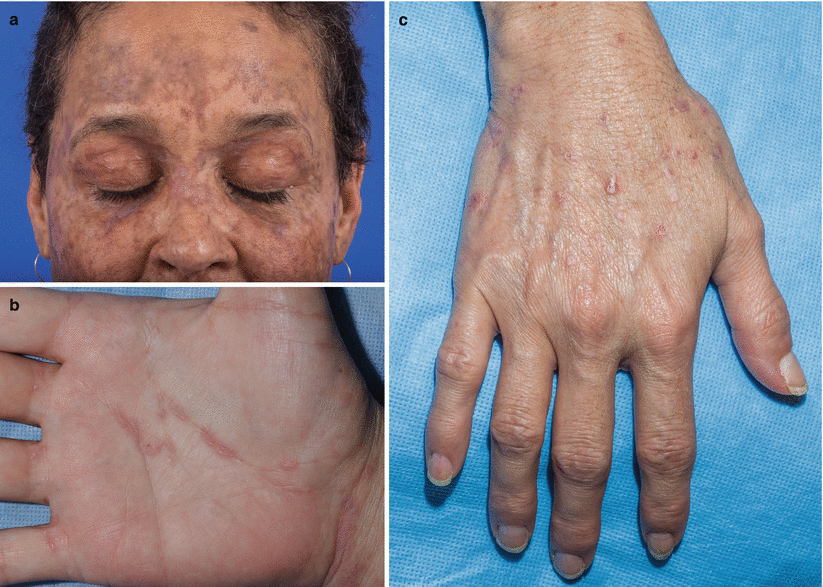
Fig. 10.2
Lichen planus–like chronic graft-versus-host disease (cGVHD). (a) Woman with lichen planus–like cGVHD on the face. (b) Man with lichen planus–like cGVHD on the palm and wrist. (c) Woman with lichen planus–like papules on the dorsal hand and wrist
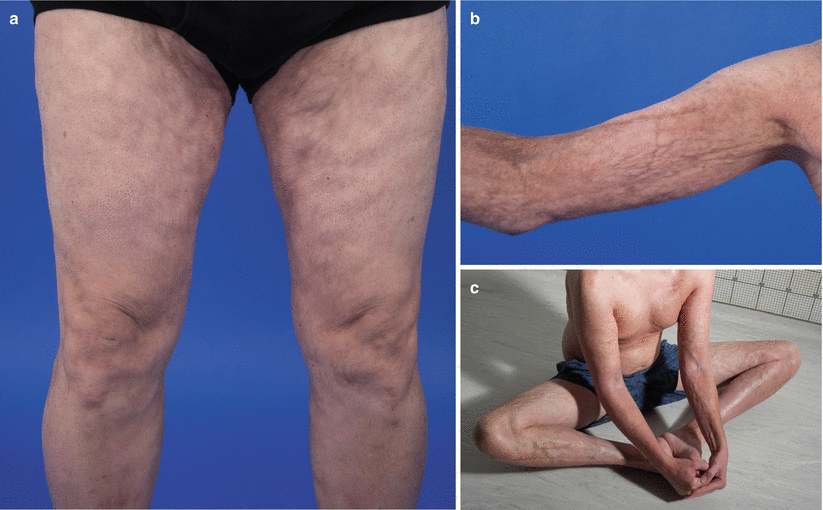
Fig. 10.3
Deep sclerosis. (a) Rippling of the subcutaneous tissue of the medial thighs from subcutaneous sclerosis. (b) Rippling and nodularity of the subcutaneous tissue of the upper extremity from subcutaneous sclerosis. (c) Rippling and nodularity of subcutaneous tissue of the upper and lower extremities accentuated by abduction and flexion of the hip
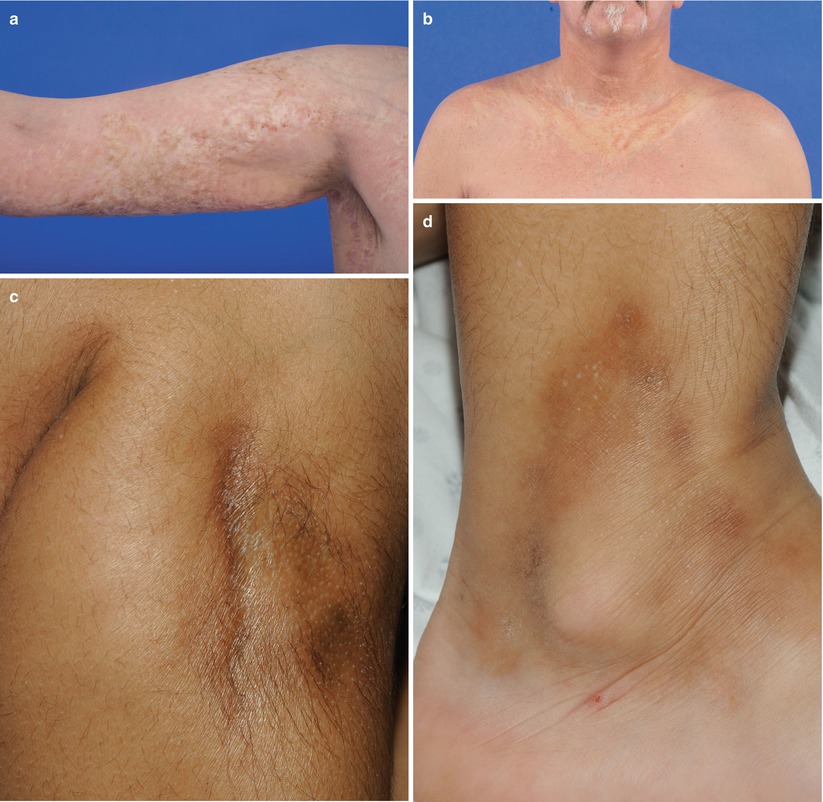
Fig. 10.4
Dermal sclerosis. (a) Thin, yellow morphea-like plaques and hyperpigmentation on the upper extremity. (b) Thin, yellow morphea-like plaques on the upper chest and anterior neck. (c) Bound down, hyperpigmented morphea-like plaque on the right leg. (d) Bound-down, hyperpigmented morphea-like plaque on right lower leg
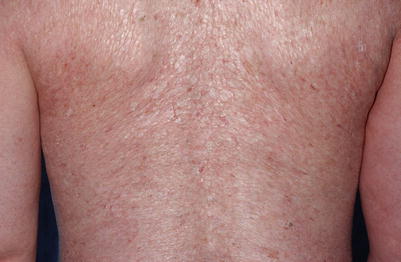
Fig. 10.5
Diffuse, thin, shiny lichen sclerosus–like plaques on the back
Diagnostic signs and symptoms that do not require additional diagnostic information to make the diagnosis of cGVHD
Distinctive signs and symptoms, which are known characteristics of cGVHD but are not sufficient to make the diagnosis of cGVHD without additional testing
Nondiagnostic features, which are controversial or rare
Signs and symptoms that are common to both acute and chronic GVHD
Table 10.1
NIH consensus project classification of signs and symptoms of cutaneous cGVHD
Site | Diagnostic | Distinctive | Other | Common |
|---|---|---|---|---|
Skin | Poikiloderma Lichen planus–like Sclerotic features Morphea features Lichen sclerosus–like | Depigmentation Papulosquamous lesions | Sweat impairment Ichthyosis Keratosis pilaris Hypopigmentation Hyperpigmentation | Erythema Maculopapular rash Pruritus |
Nails | Dystrophy Longitudinal ridging, splitting, or brittle features Onycholysis Pterygium unguis Nail loss | |||
Scalp and body hair | New-onset scarring or nonscarring alopecia Scaling | Thinning scalp hair Premature gray hair |
Diagnosis of cutaneous cGVHD requires at least one clinical diagnostic feature or at least one distinctive clinical manifestation plus a skin biopsy demonstrating cGVHD. Nondiagnostic and common features of cutaneous cGVHD are not considered for the diagnosis of cGVHD, but they are included in the severity scoring, as discussed in the following section.
Classification of cGVHD severity is also guided by the Diagnosis and Staging Working Group report. Based on the 2014 report [3], two individual skin scores comprise the overall cGVHD severity (Table 10.2). The body surface area (BSA) involvement of five cutaneous findings (maculopapular rash/erythema, lichen planus–like features, sclerotic features, papulosquamous lesions or ichthyosis, and keratosis pilaris–like GVHD) is scored from 0 to 3 based on the percentage of body surface area involved. The second skin score is based on skin sclerosis. The presence of superficial sclerosis results in a score of 2 and presence of deep sclerosis, impaired mobility, or ulceration is given a score of 3. Six other features are listed on the screening document but do not contribute to the severity score. The highest of the two skin scores is used for the NIH Global Severity Score. The global score defines severity of cGVHD, with a skin score of 1 corresponding to mild cGVHD, 2 corresponding to moderate cGVHD, and three corresponding to severe cGVHD.

Table 10.2
NIH consensus project severity scoring of cutaneous cGVHDa
Treatment
It has been estimated that approximately 50 % of patients with cGVHD have resolution of symptoms within 7 years of initiation of systemic management, and approximately 10 % will require continued systemic treatment beyond 7 years. The remaining 40 % either die or have a relapse of malignancy within 7 years of initiating treatment for cGVHD [4]. Anticipating protracted systemic therapy, many factors must be considered when selecting therapies, including disease severity, preservation of the graft-versus-tumor effect, drug toxicity, other medical comorbidities, access to therapy, and drug interactions.
Treatment of Mild Cutaneous cGVHD
Symptomatic mild cutaneous cGVHD is generally treated locally with topical steroids or topical calcineurin inhibitors (CNIs) [5, 6]. Topical therapy minimizes systemic side effects and does not interfere with the graft-versus-tumor effect. Close monitoring for disease progression is important, as many patients will eventually require systemic therapy. In addition, active skin involvement should heighten surveillance for de novo development of cGVHD in other organs. Steroids of mid to high potency can be used for erythematous and scaling lesions [7]. The topical CNIs, tacrolimus and pimecrolimus, are recommended for treatment of mild cGVHD in areas where high potency steroids should be limited or avoided, including the face and intertriginous regions [8]. Pimecrolimus may be better tolerated at sites of non-intact skin, although it is not as potent as tacrolimus [5, 9].
Treatment of Moderate and Severe Cutaneous cGVHD
Systemic therapy is usually indicated for moderate or severe cGVHD (cutaneous disease with a skin severity score of at least 2). A skin severity score of 2 is given for the presence of skin sclerosis or at least 19 % BSA involvement with one of the specified features. Prednisone (typically 1 mg/kg per day) has been the primary first-line therapy since the 1980s [10]. Trials to determine the optimal duration and dose of corticosteroids are lacking, but based on consensus and expert opinion, it is recommended that 1 mg/kg per day be administered for 2 weeks, followed by a slow taper [4, 6]. Prednisone dosing should be changed to an alternate-day regimen of 1 mg/kg within 6–8 weeks and maintained at that level for 2–3 months to avoid a flare of cGVHD. One recommended taper schedule is to decrease the dose by 20–30 % every 2 weeks and to return to the penultimate dose for 2–4 weeks if there is a flare of cGVHD [4].
A recently published phase II study by Solomon et al. [11] suggests an alternative to corticosteroids as first-line therapy. In this study, 25 patients with new-onset cGVHD of any organ were treated with 375 mg/m2 of rituximab once a week for 4 weeks, followed by infusions every 3 months for 1 year. Other noncorticosteroid immunosuppressive agents such as mycophenolate mofetil (MMF), tacrolimus, and sirolimus were permitted. Overall, 22 (88 %) of the patients responded to this corticosteroid-free treatment, with complete remission in 21 (95 %) of the 22 responders. Relapse of cGVHD occurred in 37 % within 24 months. This small prospective trial raises the possibility of a corticosteroid-free first-line regimen with fewer toxicities for new-onset cGVHD, but these results needs to be validated in a larger, controlled trial.
Combination first-line therapy of corticosteroids plus an additional agent has been evaluated in clinical trials using cyclosporine [12], azathioprine [13], thalidomide [14], MMF [15], and hydroxychloroquine [16]. No additional benefit was demonstrated in the treatment of cGVHD, but cyclosporine in combination with prednisone resulted in less osteonecrosis than prednisone alone [12]. Based on this study, CNIs are often given in combination with corticosteroids as first-line therapy for cGVHD, in an attempt to decrease toxicity associated with prolonged corticosteroid use.
Approximately 50 % of patients will respond to corticosteroids. The remaining patients with steroid-refractory cGVHD will require second-line treatment [17]. Three criteria are generally used to define steroid-refractory cGVHD:
Progression despite 1 mg/kg per day of prednisone for 2 weeks
Stable disease after 4–8 weeks of at least 0.5 mg/kg per day
An inability to decrease the dosage of prednisone below 0.5 mg/kg per day
There is no standard second-line therapy, and limited evidence exists to guide salvage therapy. Selection of a second-line agent remains mostly trial-and-error, based on factors such as the organ systems involved, comorbidities, experience of the treating provider, and access to the treatment. The few randomized controlled trials of second-line agents in cGVHD often do not support the positive findings of case reports, case series, and small retrospective or prospective studies [15]. Therapies that have demonstrated particular benefit in cutaneous cGVHD include rituximab, extracorporeal photopheresis (ECP), phototherapy, imatinib mesylate, and mammalian target of rapamycin (mTOR) inhibitors, which are discussed in detail below. Additional salvage therapies are included in Table 10.3.
Table 10.3
Other systemic therapies reported for chronic GVHD
Drug | Results | Adverse events |
|---|---|---|
Mycophenolate mofetil | There are no randomized trials. Response rates range from 40–75 % and steroid-sparing benefits have been reported | Increased risk of relapse when used as a third immunosuppressive agent [73] |
Pentostatin | ORR in two prospective trials was 51 % [73] and 53 % [74]; skin was best-responding organ (ORR 69 %). Not recommended for pulmonary cGVHD | Nausea and infection |
Methotrexate | Pooled ORR from eight studies was 77.6 %; cutaneous GVHD had best response (42–100 %) [75] | Cytopenia and hepatic toxicity |
Hydroxychloroquine | Phase II study showed ORR of 53 % [76]. Phase III RCT did not confirm benefit [16]. | Hypertension, GI toxicity, retinal toxicity |
Clofazimine | Partial response in 55 % from one study; cutaneous GVHD had best response [77] | Skin discoloration, GI toxicity |
Thalidomide | Somnolence, constipation, neuropathy, neutropenia, venous thrombosis | |
Retinoids | ORR 74 % from one retrospective study of sclerotic GVHD [83] | Hyperlipidemia, transaminase elevation, teratogenicity, impaired wound healing |
Alemtuzumab | One phase I study showed ORR of 70 % of evaluable patients; Cutaneous and fascial GVHD had best response [84] | Infection, cytopenia |
Etanercept | Infection | |
Mesenchymal stromal cells | Infection; unclear impact on graft-versus-tumor effect | |
Azathioprine | In combination with prednisone, showed increased nonrelapse mortality and decreased overall survival [13] | Oral malignancy, infection |
Rituximab
Rituximab is a chimeric monoclonal antibody reactive with CD20, found on the cell surface of B cells. Rituximab has been proposed as a treatment for cGVHD because of the suggested role of B cells and B cell–activating factor (BAFF) in cGVHD pathophysiology, the common finding of autoantibodies in patients with cGVHD, and the similarity between cGVHD manifestations and autoimmune conditions such as Sjögren’s syndrome and systemic sclerosis [18, 19]. Prospective studies have reported overall response rates (ORR) of 63–88 % [11, 20–24]. A meta-analysis of six studies reported a pooled proportion ORR of 60 % for cutaneous cGVHD; the skin was the organ with the highest response rate to rituximab therapy [25]. Regimens can vary, but patients usually receive a minimum of four weekly rituximab infusions of 375 mg/m2. Additional infusions at intervals of 1 to 3 months have also been given. Van Dorp et al. [22] studied the immunologic phenotype of 18 patients with steroid-refractory or steroid-dependent cGVHD who were treated with rituximab to evaluate predictors of response to therapy. The ORR of rituximab for skin cGVHD was greater than 75 % regardless of the type of cutaneous involvement. About three fourths of the patients were able to decrease or discontinue corticosteroids. No correlation was seen between BAFF levels and rituximab response, but an elevated B-cell number prior to treatment correlated with response to therapy with rituximab [22].
Extracorporeal Photopheresis
Extracorporeal photopheresis (ECP) is an immunomodulatory therapy that was first reported as a treatment for GVHD in the 1980s. Since then, ECP has become a widely used therapeutic modality for cGVHD of the skin and other organs (Fig. 10.6) [27–29]. As with other interventions, published reports are primarily retrospective in nature. One multicenter, randomized controlled trial compared standard treatment plus ECP (weekly on two consecutive days) to standard treatment alone over 12 weeks, for treatment of cutaneous cGVHD. The primary endpoint, a lower total skin score (TSS), had a positive trend, but it did not reach statistical significance. However, there was a steroid-sparing effect in the group that received ECP. Patients in the ECP arm were more likely to achieve both a reduction in steroid dose greater than 50 % and a total daily dose of 10 mg or less. The difference in the reduction of the steroid dose became apparent after week 8, though improvement in the TSS was noted earlier [30]. A steroid-sparing benefit of ECP also has been demonstrated in other studies [31–33].
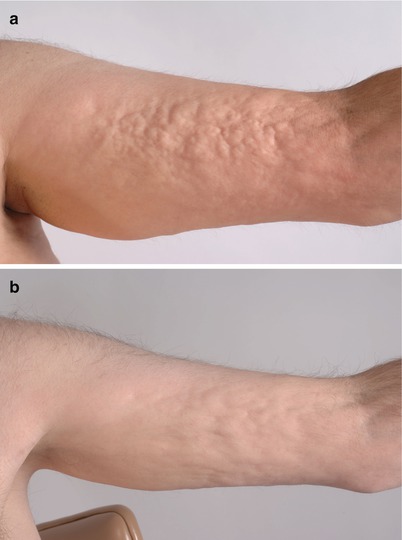
Fig. 10.6
Clinical improvement of sclerosis from extracorporeal photopheresis (ECP) therapy. (a) Localized rippling and nodularity from subcutaneous cGVHD on the left arm. (b) Decreased nodularity after 6 months of ECP
As with other salvage therapies for cGVHD, comparison of the published literature for ECP is limited because of the heterogeneity of the disease and organ-specific outcome measures, as well as the variability in the schedule of ECP administration. Nonetheless, the skin appears to be among the organs most responsive to this treatment modality. A meta-analysis of ECP for steroid-refractory cGVHD found that ECP has the greatest benefit in skin, oral, liver, and musculoskeletal disease, with pooled response rates of 74 %, 72 %, 68 %, and 64 % respectively. Response rates were lower for other organs [32]. Similarly, a systematic review of ECP for acute or chronic steroid-refractory or steroid-dependent GVHD found a pooled ORR of 71 % for cutaneous chronic GVHD, which was higher than for other target organs with cGVHD [31].
ECP is generally a safe therapeutic option for cGVHD. The most commonly reported AEs are nausea, headache, and fever. Serious infections, including line infection, are rare, and relapse of malignancy has not been reported [27, 33, 34]. Both the U.K. Photopheresis Expert Group [33] and the Ontario Stem Cell Transplant Steering Committee [35] have developed consensus statements that recommend ECP as second-line therapy for cGVHD. The U.K. consensus statement recommends delivery of ECP on two consecutive days every other week, tapering to every 4 weeks once a response is seen. At this time, there is no biologic marker of disease response, so clinical assessment is the standard outcome measure. The clinical response of cGVHD to ECP is slow; 4 months is the recommended timeframe for response assessment [33].
Phototherapy
Treatment of skin disease with ultraviolet (UV) radiation is commonly used for dermatologic conditions. As with many cGVHD therapies, high-quality trial data on the use of phototherapy is rare. In addition, different types of disease (e.g., lichenoid vs sclerotic) would be expected to have different responses, given the therapeutic penetration of UV light. No prospective studies have been carried out to provide recommendations for the type, dose, or schedule of UV therapy. Psoralen plus ultraviolet A (PUVA) is the best-studied phototherapy in cGVHD. In a retrospective analysis of 40 patients with cGVHD, 77 % had a local response (cutaneous or oral), with 40 % experiencing a complete response [36]. However, all but one of the patients included had concomitant systemic therapy. UVA1, a subset of the UVA wavelength (340–400 nm), has been evaluated in three small studies [37–39]. Overall, 24 of 25 total patients in the three studies responded, including those with both lichenoid and sclerotic cGVHD. Calzavara Pinton et al. [37] reported that all patients with lichenoid cGVHD relapsed within a month and required maintenance phototherapy. Only two studies have evaluated UVB (280–320 nm) as a local therapy for cGVHD [40, 41]. Brazzelli et al. [41] administered narrow band UVB (311–312 nm) to 10 pediatric patients, 5 with cGVHD and 5 with overlap GVHD. Of the 10 patients, 8 had a complete response, which was sustained in 71 % of the responders at 2 years.
Phototherapy is generally well tolerated. Photosensitizing medications (e.g., voriconazole, trimethoprim-sulfamethoxazole, and furosemide) should be discontinued prior to initiating phototherapy. Erythema is a common short-term AE. Oral psoralens may induce nausea, vomiting, dizziness, and hepatotoxicity, which can limit tolerability. Bath PUVA avoids the systemic side effects of psoralens, but is time-intensive. Strict sun protection (including eye protection) is required on the day psoralens are taken. Long-term phototherapy may cause accelerated photoaging and increase the risk of skin cancer, particularly with PUVA. Patients treated with PUVA develop squamous cell carcinoma more often than basal cell carcinoma or melanoma [42].
Imatinib Mesylate
Imatinib is a tyrosine kinase inhibitor (TKI) that targets the platelet-derived growth factor receptor (PDGFR) among other tyrosine kinases [43]. Stimulatory autoantibodies to the PDGFR were reported in patients with sclerotic cGVHD and in patients with systemic sclerosis, suggesting a potential pathway of skin fibrosis targetable by this drug [44, 45]. In addition, in vitro studies have demonstrated that treatment of fibroblasts with imatinib inhibited proliferation [46]. Clinical studies have reported an ORR in sclerotic GVHD ranging from 26 % to 63 % [47–52]. Comparisons between studies is challenging because of the heterogeneous disease manifestations and the range of end points used to determine efficacy. Even within studies, the benefit of this antifibrotic therapy was not consistent among patients with sclerotic GVHD.
Imatinib appears to be less well tolerated in the cGVHD setting than in chronic myeloid leukemia (CML) and other indications. AEs have been reported at doses as low as 100 mg in sclerotic GVHD patients, despite dosing of 400 to 800 mg in the CML setting [51]. Hematologic AEs include neutropenia, thrombocytopenia, leukopenia, and anemia. Nonhematologic AEs include fatigue, nausea, diarrhea, electrolyte abnormalities, fluid retention (peripheral and periorbital), and muscular cramps. Sclerotic cGVHD patients are particularly sensitive to discomfort from fluid retention and muscular cramps, as they often experience these symptoms as part of their primary disease.
Dasatinib, a second-generation TKI, has been evaluated as an alternative agent in patients with sclerotic cGVHD who are resistant or intolerant to imatinib. Three patients with sclerotic cGVHD who could not take imatinib were treated with dasatinib [53]. All three achieved a partial response and corticosteroids were decreased in all patients. In contrast to imatinib, the patients did not experience dose-limiting toxicities from dasatinib. There is overlap in the observed AEs between imatinib and dasatinib, but it appears that individual patients develop differing AEs to the two different drugs [54]. This limited experience in cGVHD suggests that dasatinib warrants further investigation as a better-tolerated treatment for some patients with sclerotic cGVHD.
Mammalian Target of Rapamycin (mTOR) Inhibitors
Sirolimus and its derivative everolimus are macrolide antibiotics that act through inhibition of the mTOR pathway. They act as immunosuppressive drugs and may also act via inhibition of proliferation of fibroblasts and smooth muscle cells. Additionally, they have a role in reconstitution of regulatory T cells in cGVHD [55]. mTOR inhibitors have been suggested as alternatives to CNIs for treatment of cGVHD because of the protective effect on skin cancer development that has been demonstrated in recipients of solid-organ transplants [56]. Data regarding the efficacy of mTOR inhibitors specifically for cutaneous cGVHD are scarce. One prospective study reported a response rate of 65 % in skin cGVHD, and one retrospective study of sclerotic cGVHD reported an ORR of 76 % [57, 58]. Additionally, mTOR inhibitors have demonstrated a steroid-sparing benefit.
The combination of CNIs and mTOR inhibitors is associated with increased risk of transplant-associated microangiopathy and renal toxicity [57]. Other toxicities seen with mTOR inhibitors include hyperlipidemia, infection, and cytopenias. mTOR inhibitors are substrates for cytochrome p450 3 A4, indicating that potential drug interactions may occur in GVHD patients, who often take multiple medications. Potential impairment of wound healing should also be considered for patients with skin breakdown or ulceration [59].
Related posts:
 Grading and Treatment of Acute Graft-Versus-Host Disease
Grading and Treatment of Acute Graft-Versus-Host Disease
 Wound Care in the Management of Chronic Graft-Versus-Host Disease
Wound Care in the Management of Chronic Graft-Versus-Host Disease
 Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
 Dermal and Subcutaneous Chronic Graft-Versus-Host Disease
Dermal and Subcutaneous Chronic Graft-Versus-Host Disease
 Diagnosis, Staging, and Treatment of Chronic Graft-Versus-Host Disease
Diagnosis, Staging, and Treatment of Chronic Graft-Versus-Host Disease
 Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
Clinical Presentation of Nonsclerotic Epidermal Chronic Graft-Versus-Host Disease and Hair and Nail Changes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


