Key Points
- ▪
Lymphedema occurs as a result of congenital abnormalities (primary lymphedema), injury or infection (secondary lymphedema) of the lymphatic system.
- ▪
Most common cause in developed countries is cancer treatment.
- ▪
Secondary upper limb lymphedema develops on average, eight months after axillary lymphadenectomy.
- ▪
77% of patients who go on to develop breast cancer-related lymphedema do so within three years of surgery.
- ▪
Diagnosis of lymphedema is based on history, physical exam, physiologic, and radiologic measures.
- ▪
The pathophysiology of lymphedema remains poorly understood, but involves changes that promote fibrosis and adipose deposition.
- ▪
Obesity, radiation and infection are significant risk factors for secondary lymphedema.
Introduction
Lymphedema is a complex clinical condition that is characterized by edema, fibrosis and adipose deposition. Lymphedema may occur as a result of genetic or developmental abnormalities (primary lymphedema), or as a consequence of postnatal insults such as trauma, radiation, or infection (secondary lymphedema). It is estimated that 140 to 250 million people worldwide suffer from this disease with the vast majority of cases occurring in developing countries secondary to parasitic infections by Wuchereria bancrofti . In Westernized countries, lymphedema most commonly occurs after cancer treatment. Due to the high incidence of breast cancer, survivors of this disease constitute the largest number of patients with postsurgical lymphedema. It is estimated that 30–50% of patients who undergo axillary lymph node dissection for breast cancer management go on to develop lymphedema. Even trivial lymphatic injury in the form of sentinel lymph node biopsy has been shown to be associated with lymphedema development in 5–7% of patients. However, development of secondary lymphedema is not limited to breast cancer survivors; in fact, it is estimated that as many as one in six patients treated for a variety of other solid malignancies (most commonly melanoma, gynecological tumors, and sarcomas) also go on to develop lymphedema.
Natural History
Primary Lymphedema
The natural history of primary lymphedema is variable depending on the underlying developmental abnormality as well as the penetrance (i.e., severity) of the defect in the affected individual. Defective phenotypes may present immediately after birth in patients with congenital lymphedema, or in other cases may present later in life with progressive symptoms (lymphedema praecox or lymphedema tarda).
Congenital lymphedemas are thought to account for 10–25% of primary lymphedema and have a sex bias affecting females twice as commonly as males. In addition, congenital lymphedemas more commonly involve the lower extremities rather than the upper extremities. A small subset of congenital lymphedema (∼2% of all patients) suffer from Milroy’s disease, which is a familial, sex-linked disorder caused by loss-of-function mutations in the vascular endothelial growth factor-3 receptor gene. These patients have hypoplastic lymphatics and variable degrees of dermal and collecting lymphatic vessel agenesis.
Patients with lymphedema praecox typically present with unilateral lower extremity lymphedema (∼70%) at some point after birth and before age 35. These patients characteristically have a decreased number and caliber of lymphatics and most commonly present during puberty with a female to male ratio of 4:1.
Finally, lymphedema tarda is a primary lymphedema that manifests clinically after the age of 35. These cases represent only a minority of patients with primary lymphedema (<10%) and, similar to other forms of primary lymphedema, most commonly affect the lower extremity of women. In general, lymphedema tarda is a diagnosis of exclusion although recent studies have demonstrated loss of function mutations in the FOXC2 (forkhead box C2) gene.
Recent advances in molecular biology and genetic sequencing have revolutionized diagnosis of primary lymphedema and have led away from the scheme presented above. These advances have identified specific genetic mutations and have the promise of improving our understanding of these disease processes with development of targeted treatments. Genetic mutations resulting in lymphatic abnormalities are summarized in Table 6.1 .
| Gene | Syndrome | Associated Conditions | Reference |
|---|---|---|---|
| FLT4 (5q35) (encodes for VEGFR3, vascular endothelial growth factor receptor 3) | Milroy disease | Primary lymphedema and prominent vascularity in feet, upslanting ‘ski jump’ toe nails due to edema of nail bed | |
| FLT4 (4q34) (encodes for VEGFC, vascular endothelial growth factor C) | Milroy-like lymphedema | Congenital lymphedema in lower limbs | |
| FOXC2 | Lymphedema-distichiasis syndrome | Lower-limb lymphedema at puberty, development of a double row of eyelashes | |
| SOX18 | Lypotrichosis-lymphedema- telangiectasia syndrome | Lymphedema coupled with hair loss and small dilated blood vessels near the surface of skin | |
| IL-6 (Interleukin 6) | No name | Loss of IL-6 results in increased localized adiposity near damaged lymphatics | |
| GJC2 (encodes for CX47, connexin 47) | Meige disease | CX47 mutations are associated with impaired gap junction activities that disrupt lymphatic flow. Mutations of CX47 have also been suggested to predispose patients undergoing breast cancer treatment therapy to secondary lymphedema | |
| CCBE1 (18q21) (encodes for collagen & calcium binding EGF domain 1) | Hennekam syndrome | Severe lymphedema in limbs, genitalia, and face, facial anomalies, seizures, mental retardation, and stunted growth. Symptoms often present in utero | |
| KIF11 (10q24) | MCLMR syndrome | Microcephaly, congenital lower limb lymphedema, ocular problems, mild to moderate learning disabilities | |
| GATA2 (3q21) | Emberger syndrome WILD syndrome | Lower limb lymphedema in either one or both legs in childhood, severe cutaneous warts, myelodysplasia GATA2 is also thought to contribute to WILD syndrome (warts, immunodeficiency, lymphedema, and dysplasia) | |
| AKT1 | Proteus syndrome | Progressive, segmental overgrowth of skin, skeleton, connective tissue, and CNS, lymphatic malformations |
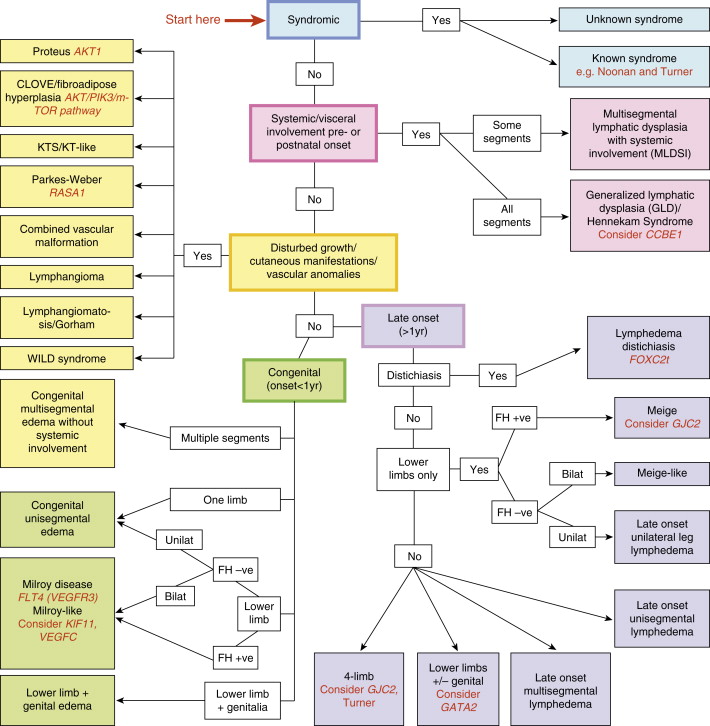
As noted above, primary lymphedemas have traditionally been subclassified by their time of onset into these three general groups; however, the presentation of symptoms varies greatly within these categories. A review published by Connell et al. describes a more detailed classification system that is not only based on the presentation of symptoms (such as timing of onset and affected limb), but also distinguishes between many of the specific genetic factors outlined in Table 6.1 . This classification system categorizes congenital lymphedemas into five main groups: syndromic, systemic or visceral, disturbed growth, congenital onset, and late onset ( Figure 6.1 ). This system can be used as a diagnostic algorithm for delineating the many specific phenotypes of primary lymphedema in a more precise manner.
Secondary Lymphedema
Secondary lymphedema is caused by injury or obstruction of the lymphatic system. The most common cause of secondary lymphedema worldwide is filariasis, a disease in which parasitic roundworms occupy and occlude lymphatic vessels. This disease process has been extensively studied epidemiologically and pathologically; however, this discussion is beyond the scope of this chapter.
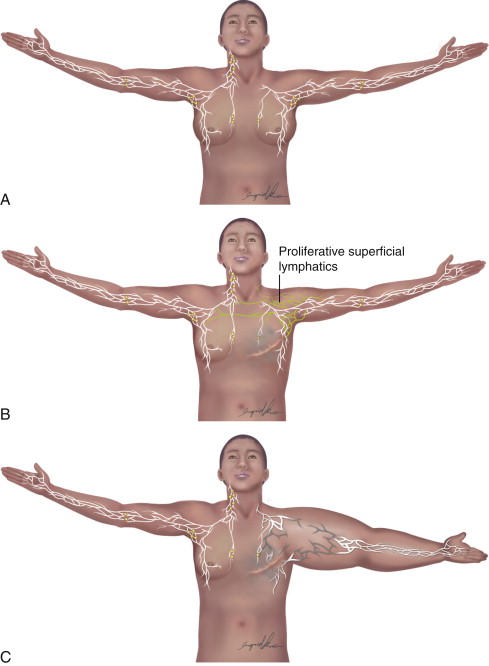
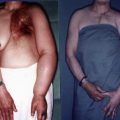
In developed countries, secondary lymphedema occurs most frequently as a consequence of cancer treatment. Although the majority of published literature on cancer-related lymphedema involves breast cancer survivors, lymphedema is also a common complication of other solid tumors. In most cases, the development of lymphedema after surgery occurs in a delayed fashion, presenting months and sometimes even years after initial treatment ( Figure 6.2 ). Recent reviews have shown that breast cancer-related lymphedema presents, on average, approximately eight months after axillary lymph node dissection, and more than 75% of those who eventually develop lymphedema do so within the first three years postoperatively. Thus, although many patients develop acute postoperative edema of the upper extremity, this edema typically resolves over a period of weeks postoperatively only to reoccur more permanently at a later time. In addition, although there is less information regarding the timing of lower extremity lymphedema development after surgery, the available evidence suggests that these patients present somewhat earlier than upper extremity cases (but still rarely beginning immediately after surgery unless a massive soft tissue resection and lymphatic ablation is performed).
Progression of secondary lymphedema, once established, is highly variable, with some patients experiencing relatively indolent disease requiring no extensive physical therapy or compression treatments, and others rapidly progressing to disabling disease that interferes with activities of daily living even despite these interventions. In general, however, lymphedema tends to be progressive unless palliative measures such as manual lymphatic massage and compression garments are undertaken. There is some debate, however, about how often and the extent to which these interventions should be performed.
Recently, a number of studies have shown that exercise and weight loss regimens are effective means of preventing progression of, and in some cases even treating lymphedema. These results, when first published, were revolutionary, since prior to formal studies patients who were at risk for lymphedema or had established lymphedema were routinely warned to avoid exercise; this was based on the fear that increasing the blood flow to the affected extremity would increase interstitial fluid load and thereby increase the risk/severity of lymphedema. However, since their initial description, a number of level I, prospective controlled randomized studies have proven this dogma incorrect and it is clear that exercise is a helpful modality in this population.
Diagnosis
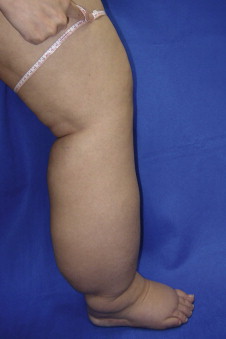
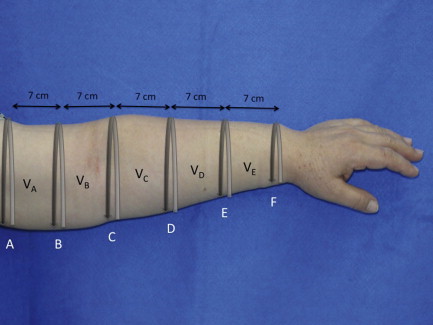
Lymphedema is diagnosed by history, physical examination and physiologic measures. The common symptoms of lymphedema include limb swelling, skin tightness, heaviness, fatigue, and skin infections. Limb circumference or volume changes relative to the normal limb can be used as a means of analyzing differential changes; typically, a difference in circumference of more than 2 cm ( Figure 6.3 ) or a volume differential of greater than 200 cc ( Figure 6.4 ) is considered significant. Often these patients also have a history of lymphatic injury, cancer treatment and/or radiation. A family history may also be present in patients presenting with primary lymphedema. The differential diagnosis for primary or secondary lymphedema includes venous insufficiency, congestive heart failure, malignancy (either recurrent or primary causing lymphatic obstruction) and infections.
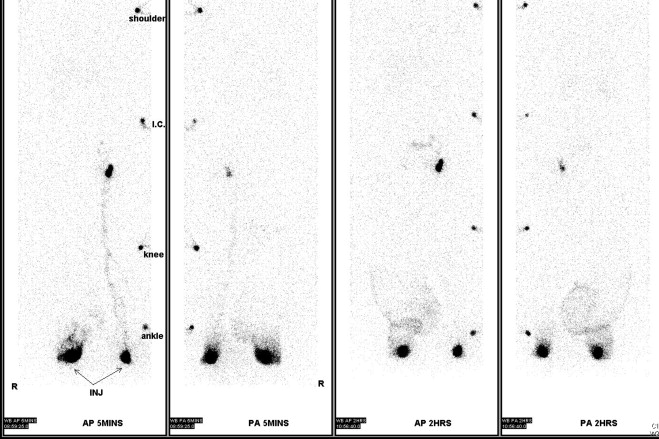
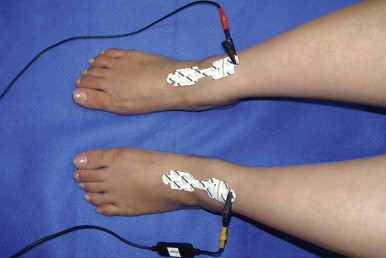
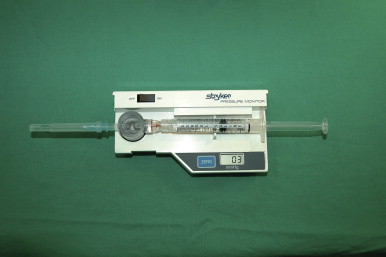
A number of tests can be used to aid in the diagnosis of lymphedema. Lymphoscintigraphy involves injection of a radiolabelled colloid in the distal limb and analysis of proximal lymph node uptake. These studies are, in general, performed as a qualitative analysis (i.e., presence or absence of lymph node uptake; Figure 6.5 ); however, some centers have reported quantitative lymphoscintigraphic analysis based on decay-adjusted uptake and lymphatic transport index. Similarly, magnetic resonance imaging (MRI) lymphangiography has been reported but has not been widely adopted. Noninvasive methods can also be used to help diagnose lymphedema. For example, a perometer can be used to calculate limb volumes (more accurate than limb circumference) using infrared scanning to measure the circumference of multiple areas of the limb. Bioimpedence technologies calculate the rate of electrical current transmission through tissues and have been used to estimate the fluid content of the limb as compared with the normal limb ( Figure 6.6 ). This test has been shown to be particularly helpful with early-stage lymphedema. Skin tonometry ( Figure 6.7 ) can be used to measure skin and soft tissue fibrosis, a hallmark of lymphedema.
Staging
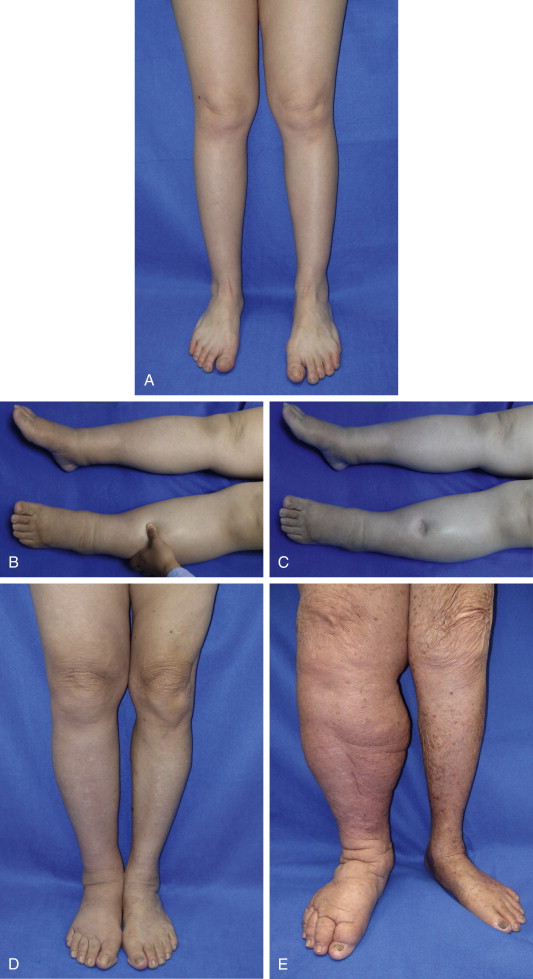
A number of classification schemes based on clinical features and changes in limb swelling have been proposed. The International Society of Lymphology staging system is the most widely used and is illustrated in Figure 6.8 and summarized in Table 6.2 . Briefly, patients are classified as having stage 0 lymphedema (latent) when lymphatic vessels have been injured resulting in impairment of fluid transport, but have no measurable swelling or edema. Stage I lymphedema (spontaneously reversible lymphedema) occurs with the onset of measurable swelling and pitting of the skin and improvement in these symptoms with elevation or compression garments. When significant fatty acid deposits and protein-rich fluid accumulation prevent compression treatment from being a viable method to reduce symptoms, the patient is considered to be at stage II (spontaneously irreversible lymphedema). Stage III lymphedema, lymphostatic elephantiasis, is the final and most severe stage of the progression of lymphedema. It is characterized by severe swelling, fibrosis, adiposity and significant thickening of skin in the form of hyperkeratosis or acanthosis. Campisi et al. have proposed a similar staging scheme with stage I defined as initial or irregular edema, stage II, as persistent lymphedema, stage III, as persistent lymphedema with lymphangitis, stage IV, as fibrolymphedema (‘column’ limb), and stage V, as elephantiasis.
| Stage | Term | Description |
|---|---|---|
| 0 | Latent lymphedema | Lymphatic vessels are injured. Capacity for fluid transport is impaired, but still sufficient to drain lymph as necessary. Lymphedema has not yet occurred or is not yet apparent |
| I | Spontaneously reversible lymphedema | Pitting has started – the affected area indents when pressure is applied to it. Swelling can be contained with the use of compression garments |
| II | Spontaneously irreversible lymphedema | Affected tissue is considered to be non-pitting and has a spongy consistency. Fibrosis starts to occur as the limbs harden and increase in size. At this point, compression garments are ineffective at suppressing symptoms |
| III | Lymphostatic elephantiasis | Swelling is irreversible, and the tissue is heavily fibrosed and unresponsive to treatment. Skin has become significantly thickened |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


