The approach to hematopoietic disorders in this chapter is in no small part indebted to the outstanding foundations that have been developed in previous editions of this text. In particular, the classification, images, and related commentary provided by LeBoit and McCalmont (8), Murphy and Schwarting (9), and Hsu and Murphy (10) were integral to the development of the present material and represent a major advance in the textural treatment of this difficult and rapidly evolving topic.
CUTANEOUS HEMATOPOIETIC INFILTRATES: OVERVIEW AND GENERAL CONCEPTS
Hematopoietic Cell Lineage: Relevance to Cutaneous Immune Surveillance and Lineage Differentiation
One of the major functions of skin is protection against environmental hazards, including infectious organisms. Evolution has allowed the development of a highly effective two-tiered defense mechanism composed of a rapid “innate” and a sustained “adaptive” response. The innate immunity is mediated by macrophages, leukocytes, natural killer (NK) cells, γδ T cells, and dendritic cells, which detect nonspecific epitopes commonly shared by different pathogens (such as glycolipids, lipoproteins, peptidoglycans, and double-stranded RNA) and mount immediate responses. In contrast, the adaptive arm of the cutaneous immune system targets specific protein antigens in a major histocompatibility-restricted manner through effectors like αβ T and B cells. Most cutaneous lymphomas have putative lineage derivation from and perhaps biologic behavior attributed to the distinct cell populations of the immune system and their evolutionary stages of maturation. Malignant T cells and myeloid cells tend to recapitulate morphologic and antigenic features of cells undergoing intrathymic/postthymic and intraosseous maturation, respectively. Malignant B cells also have definite developmental relationships to normal B-cell ontogeny within bone marrow and lymphoid organs.
T- and B-Cell Infiltration Patterns
While numerous exceptions exist, there are fundamental and often reproducible trends in the patterns produced when lymphocytes infiltrate the skin (Table 31-2) (11). This is based in part on differences in adhesive ligands between B and T cells, resulting in differential affinity for various compartments within the skin. Moreover, chemokinetic and chemotactic signals may influence the migration of various classes of lymphoid cells in different ways. The best example of this is the tendency for both benign and malignant T cells, but not B cells, to migrate into epithelial structures, including the epidermis and its adnexa. This results in the phenomena of “exocytosis” (generally used in benign settings) or “epidermotropism” (generally used in neoplastic settings) and “folliculotropism,” accounting for the tendency to identify T cells within the epidermal layer and often as a band of cells within the underlying papillary dermis (Fig. 31-1A). Occasionally, as is the case in epidermotropic phases of mycosis fungoides (MF), T cells may align along the basement membrane zone of the dermal–epidermal junction, a finding that may have a basis in expression by these cells of integrin receptors for the specific membrane components (12). This “T-cell pattern” of infiltration is not invariable among T-cell malignancies however, and epidermotropism may be blunted as tumors evolve and progress to more aggressive and poorly differentiated states. B cells, on the other hand, do not demonstrate the same affinity for the epidermis and adnexa, and seldom are these cells detected in significant numbers within the epidermis or papillary dermis. The sparing of the latter produces the characteristic “Grenz zone” seen in B-cell pattern infiltrates of the skin. Moreover, perhaps because of the innate proclivity of B cells to aggregate into follicular structures, B-cell infiltrates in the skin often (but not invariably) show a nodular architecture (Fig. 31-1B).
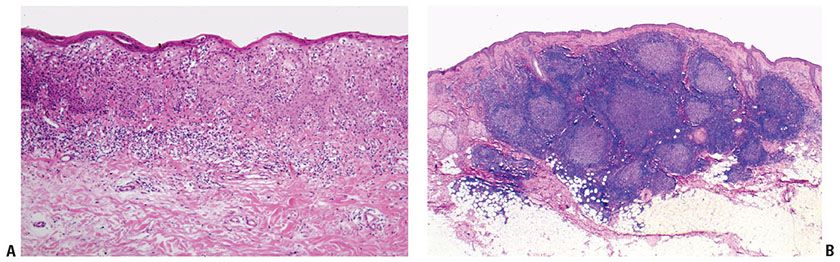
Figure 31-1 Patterns of T- and B-cell infiltration. A: T-cell pattern with infiltration of the overlying epidermis. B: B-cell pattern with Grenz zone and germinal center formation.
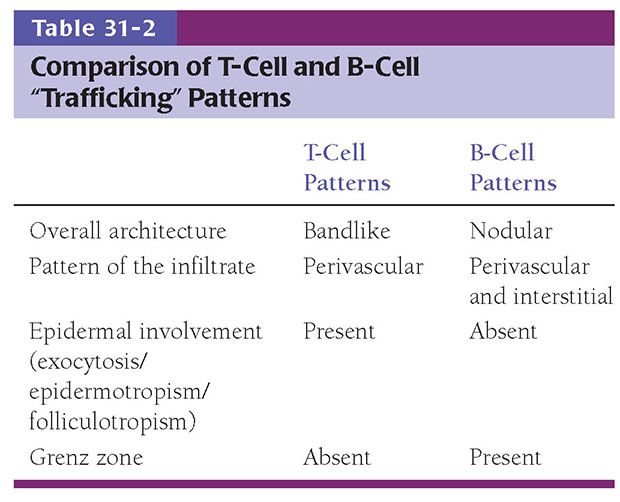
T- and B-cell patterns are potentially helpful in presumptive assignment of cell lineage, but they are not invariable, cannot be used as a reliable surrogate for immunohistochemical detection of lineage-related antigens, and do not permit separation of reactive from dysplastic and malignant populations. Although this latter distinction frequently depends on cytology, immunophenotype, and genotype, the architecture or “trafficking” patterns of hematopoietic cells in the skin may also provide insight into the biologic potential for aggressive behavior.
Abnormal Trafficking: Distinction from Inflammation
Skin is a lymphoid organ, and both the epidermal and dermal layers represent interactive immune systems that contribute to and partially regulate inflammatory cell trafficking (13,14). In both T- and B-cell responses in skin, circulating lymphocytes initially adhere to activated endothelium lining dermal postcapillary venules. Migration across the vessel wall results in perivascular or angiocentric dermatitis (Fig. 31-2A). If further migration is stunted, this exclusive angiocentric localization may produce a pattern suggestive of a specified number of differential diagnostic considerations based on correlation with clinical parameters (e.g., as in erythema chronicum migrans). Migration away from the vessel wall in other cases is presumably swift enough to permit papillary dermal accumulation of T cells and migration of T cells into the epidermis, in addition to persistent angiocentrism. However, activated T cells on an immunologic mission, as in the case of contact hypersensitivity, psoriasis, or cytotoxic dermatitis, generally alter the epidermis as they enter it by way of the cytokines and growth factors that they produce. Thus, reactive T-cell exocytosis is often accompanied by epithelial spongiosis, acanthosis, or apoptosis. In the case of malignant epidermotropic T-cell infiltration, in contrast, the epidermis tends to be “passive,” with T-cell infiltration resulting in permissive aggregation of variably atypical lymphocytes within the epidermis (Fig. 31-2B) and sometimes also or exclusively in adnexal epithelium. This infiltration is not generally associated with reactive epithelial alteration in the form of spongiosis or apoptosis but rather may be associated with epithelial injury in the form of localized mucinous degeneration, a potential contributing factor to the development of Pautrier microabscesses and follicular mucinosis that may accompany MF (15). Moreover, as the epidermis becomes more extensively infiltrated, impaired maturation often manifests as clinical abnormalities in scale production. Hence, many T-cell lymphoproliferative disorders will show surface hyperkeratosis that in early stages may clinically mimic dermatitis (Fig. 31-2C).
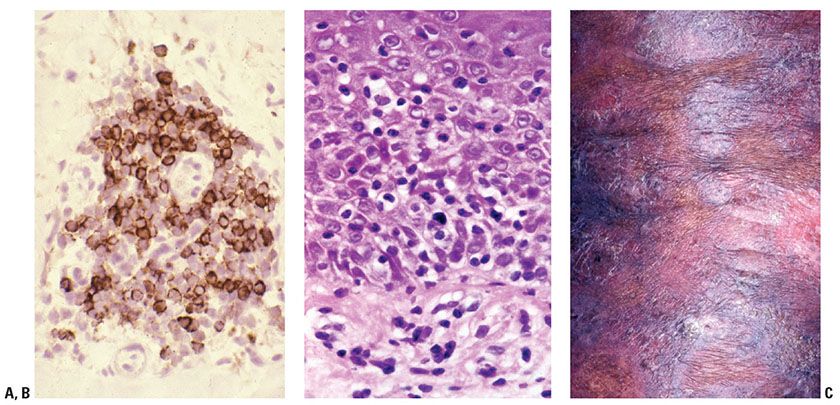
Figure 31-2 T-cell trafficking patterns. A: CD3 immunohistochemistry highlights angiocentric T cells. B: Abnormal epidermotropism into a “passive” epidermis. C: Scaling patches and plaques as a consequence of altered epidermal maturation associated with T-cell epidermotropism (MF).
Infiltration of the skin by reactive B cells often results in a perivascular accumulation of cells usually without significant involvement by the papillary dermis or epidermis, producing a Grenz zone of papillary dermal sparing and preservation of architecture. With persistence of antigenic stimulus, as may occur in the setting of some insect bite reactions, nodular aggregates of B cells may form in the dermis, recapitulating germinal centers (Fig. 31-3A). Malignant B-cell infiltrates typically show a destructive relationship to preexisting structures (Fig. 31-3B). Accordingly, malignant B cells will frequently infiltrate in an interstitial pattern (vs. perivascular), resulting in splaying apart of collagen bundles and mesenchymal cells (e.g., smooth muscle cells forming arrector pili muscles) within the reticular dermis. Moreover, nodular aggregations of malignant B cells tend to displace adnexal epithelium, resulting in their distortion and occasionally in their ischemic destruction in the process. Of note, malignant B cells may also form nodules vaguely reminiscent of germinal centers, or even induce the formation of associated reactive B-cell infiltrates.
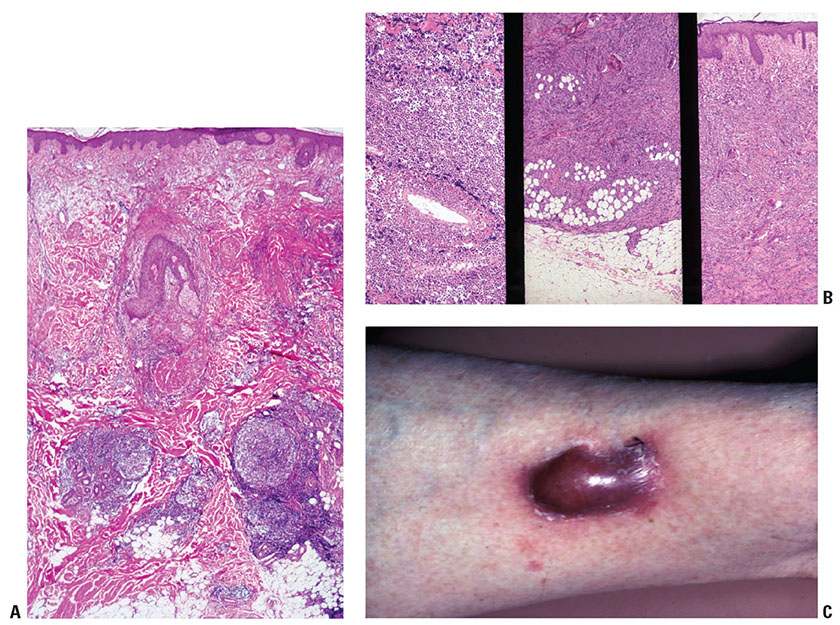
Figure 31-3 B-cell trafficking patterns. A: B-cell pattern showing nondestructive infiltration of superficial and deep dermis. B: Neoplastic B cells showing vascular invasion (left), replacement of subcutis (middle), and diffuse interstitial pattern (right). C: Plum-colored nodule covered by thinned, glistening epidermis due to B-cell dermal infiltration.
Circulating leukemia cells, like inflammatory cells, may show a perivascular pattern of infiltration on initial infiltration of skin. Although the cells avoid epithelial structures, they will evolve to a dermal interstitial pattern with time. Other nonmalignant conditions may show an interstitial architecture such as histiocytes in the setting of certain evolutionary stages of palisaded granulomatous dermatitis, lymphocytes in early inflammatory lesions of morphea, and myeloid cells in the setting of extramedullary hematopoiesis.
The tendency for progressive dermal infiltration with relative sparing of the epidermal layer in both B-cell and many leukemic malignancies results in clinical lesions that are red to plum-colored and covered by thin, shiny (nonkeratotic) epidermal surfaces (Fig. 31-3C).
PRIMARY CUTANEOUS LYMPHOMAS
Mature B-Cell Lineage
Extranodal Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissues (MALT Lymphoma)
In the 2008 WHO classification system, marginal zone lymphomas (MZL) of the skin are grouped into the broader category of extranodal MZL of mucosa-associated lymphoid tissues, also known as MALT lymphoma (1). This category encompasses MALT lymphomas arising at all extranodal sites including the gastrointestinal tract (most common), salivary gland, lung, head and neck, ocular adnexa, skin, thyroid, and breast. In the prior 2005 WHO/EORTC classification of cutaneous lymphomas, skin lesions were distinctly categorized as primary cutaneous marginal zone B-cell lymphoma (PCMZL) (2). Many still prefer this designation, and, for simplicity sake, we will use the term “PCMZL” in this chapter. This category includes plasma cell–rich variants previously designated as primary cutaneous immunocytoma, follicular lymphoid hyperplasia with monotypic plasma cells, and extramedullary plasmacytoma of the skin in the absence of underlying multiple myeloma. Among primary cutaneous lymphomas, the frequency of PCMZL is approximately 7% (2). The diagnosis of PCMZL can be very challenging owing to nonspecific morphologic features, numerous admixed reactive lymphocytes, and lack of a diagnostic immunophenotype.
Clinical Summary. Typically, PCMZL manifests as single or multiple, clustered erythematous or violaceous papules, plaques, or nodules. The lesions occur most commonly on the trunk, upper extremities, or, less commonly, in the head and neck region. Of note, head and neck lesions in older patients may be a harbinger of an underlying nodal MZL and warrant systemic investigation (16). The male-to-female ratio is approximately 2:1 with a median age of occurrence of 55, although these lesions can also arise in children (17). Plasma cell–rich variants are more commonly associated with older patients and lower-extremity lesions. In endemic areas, plasma cell–rich cases of PCMZL may be associated with the tick-associated spirochete Borrelia burgdorferi (18–24). In general, PCMZL is an indolent form of lymphoma with a 5-year survival close to 100%. Following spontaneous regression or therapy, recurrence at the same or distant sites is observed in approximately 40% of patients but typically retains a low-grade morphology. Extracutaneous spread is rare. Very rare cases of transformation to diffuse large B-cell lymphoma (DLBCL) have been reported (25).
Histopathology. In PCMZL, the neoplastic lymphocytes form a nodular or diffuse dermal infiltrate, sparing the overlying epidermis (Fig. 31-4A, B). The infiltrate often surrounds reactive (nonneoplastic) follicles, which may contain germinal centers (Fig. 31-5A, B). Unlike other extranodal MALT lymphomas, lymphoepithelial lesions are not a feature of PCMZL. The infiltrate may extend into the underlying subcutaneous tissue. The cells have small- to medium-sized nuclei with slightly irregular contours and inconspicuous nuclei and thus resemble centrocytes (Fig. 31-6A). The cytoplasm is pale; cells showing less cytoplasm resemble small lymphocytes, while those with more ample cytoplasm acquire a monocytoid appearance (Fig. 31-6B). Variable plasmacytic differentiation may also be present in the form of lymphoplasmacytoid cells or plasma cells (Fig. 31-7A), and in these cases Dutcher bodies (pale nuclear pseudoinclusions) or intracytoplasmic Russell bodies may be present and help to signify neoplasia (Fig. 31-7B). Reactive plasma cells are often found at the periphery of the infiltrate. A minority population of centroblasts and immunoblasts, sometimes quite large in size, may also be observed. Many reactive T and B cells may be present and may outnumber the neoplastic B cells, confounding interpretation (26).
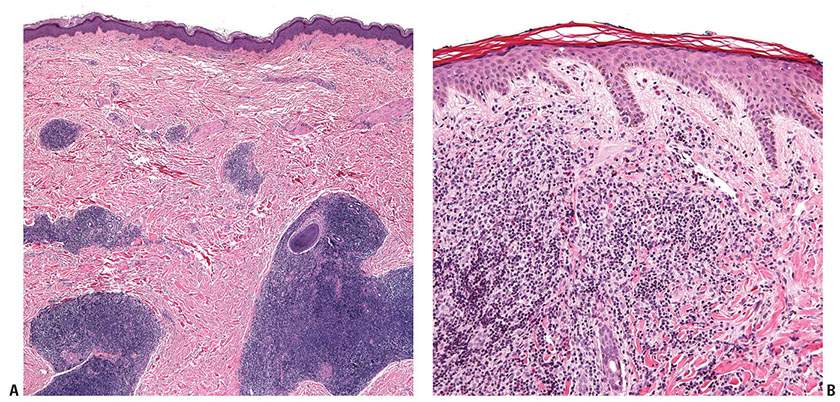
Figure 31-4 Primary cutaneous marginal zone lymphoma. A: Nodular infiltrate of neoplastic B cells forming dense, irregularly shaped aggregates in the reticular dermis. B: Diffuse dermal infiltrate of small-sized neoplastic B cells, separated from the overlying epidermis by a Grenz zone.
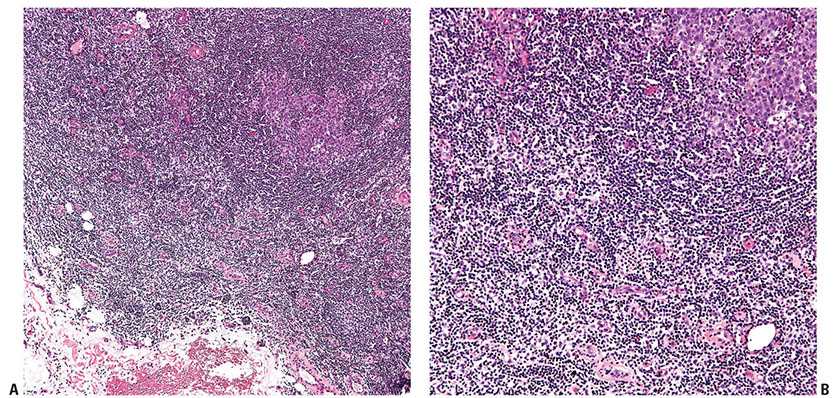
Figure 31-5 Primary cutaneous marginal zone lymphoma. A: Low-magnification view of a reactive germinal center and associated mantle zone surrounded by a neoplastic lymphoid infiltrate. B: Higher magnification showing the larger cells of the germinal center (upper right corner), the surrounding concentric layers of the mantle zone, and the interstitial neoplastic infiltrate.
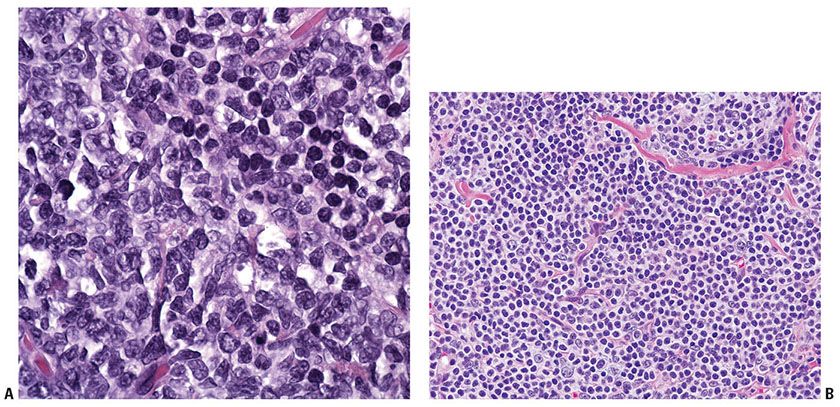
Figure 31-6 Primary cutaneous marginal zone lymphoma. A: The atypical neoplastic cells have irregular nuclear contours and slightly dispersed chromatin, compared to the admixed nonneoplastic lymphocytes with round nuclear contours and condensed chromatin (seen in the upper part of the field). B: Some proliferations are composed of cells with abundant clear cytoplasm, imparting a “monocytoid” appearance.
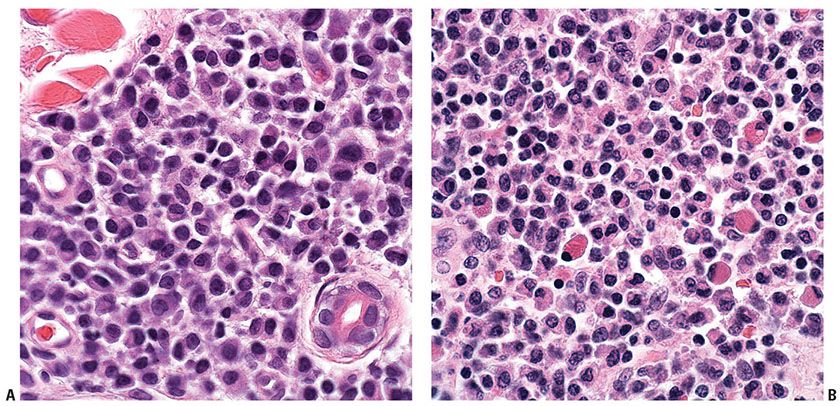
Figure 31-7 Primary cutaneous marginal zone lymphoma. A: Morphologic evidence of plasmacytoid differentiation as evidenced by the presence of eccentrically placed nuclei and, in some cells, a perinuclear clearing or “hof.” B: Large, eosinophilic globules (“Russell bodies”) are seen in the cytoplasm of some cells engorged with immunoglobulin.
Tumor cells show immunoglobulin light chain restriction. In the modern era, this is best documented by flow cytometry, although in situ hybridization for mRNA may be helpful if monotypic lymphoplasmacytoid forms or plasma cells are admixed (Fig. 31-8A, B). Importantly, in situ hybridization studies are only successful in cells with abundant cytoplasmic immunoglobulin mRNA (i.e., plasma cells or lymphoplasmacytoid cells). Flow cytometry may be falsely negative if the number of reactive B cells masks a minor clonal population; similarly, a reactive plasma cell population may overshadow a neoplastic plasma cell infiltrate by in situ hybridization. The PCMZL B cells will be positive for CD20, but CD79a may be needed to pick up the lymphoplasmacytoid/plasma cell forms. While a dense population of B cells at an extranodal site is a good clue that the infiltrate is neoplastic, no specific marker exists to confirm the diagnosis of PCMZL. The B cells express BCL2 and do not express CD5, CD10, and BCL6 (Fig. 31-9A–F). Unlike other MALT lymphomas, aberrant CD43 expression is not commonly found (Fig. 31-10A, B). CD21 will highlight disrupted follicular dendritic cell meshworks.
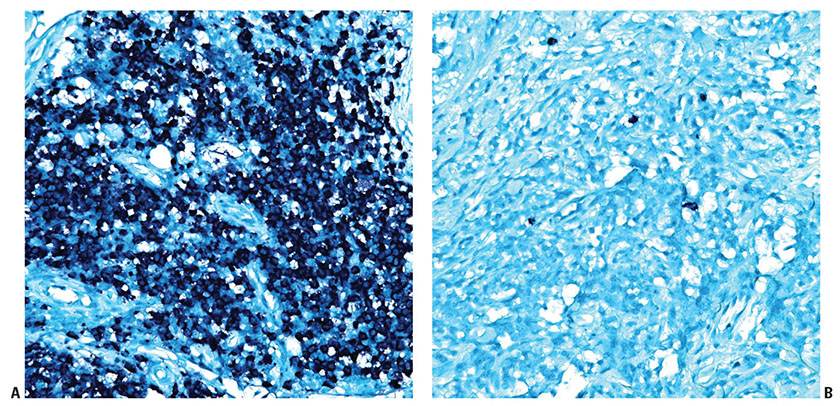
Figure 31-8 Primary cutaneous marginal zone lymphoma. A: In situ hybridization for cytoplasmic immunoglobulin kappa light chain mRNA demonstrates a kappa-monotypic population. B: In situ hybridization for cytoplasmic immunoglobulin lambda light chain mRNA in the same case reveals only rare lambda-restricted plasma cells, which represent few admixed reactive (nonneoplastic) plasma cells.
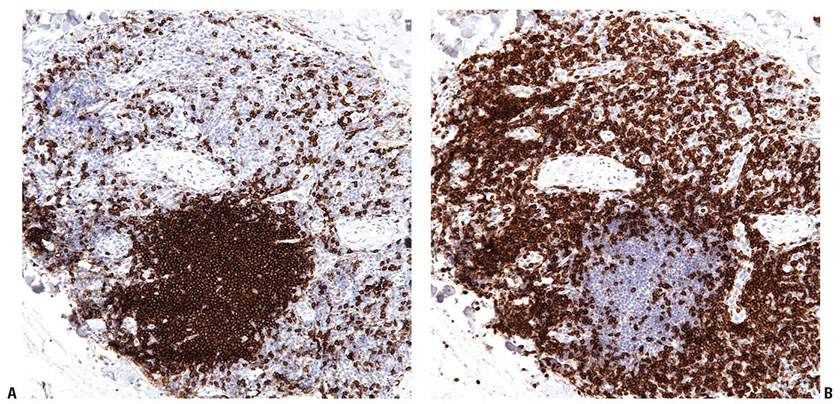
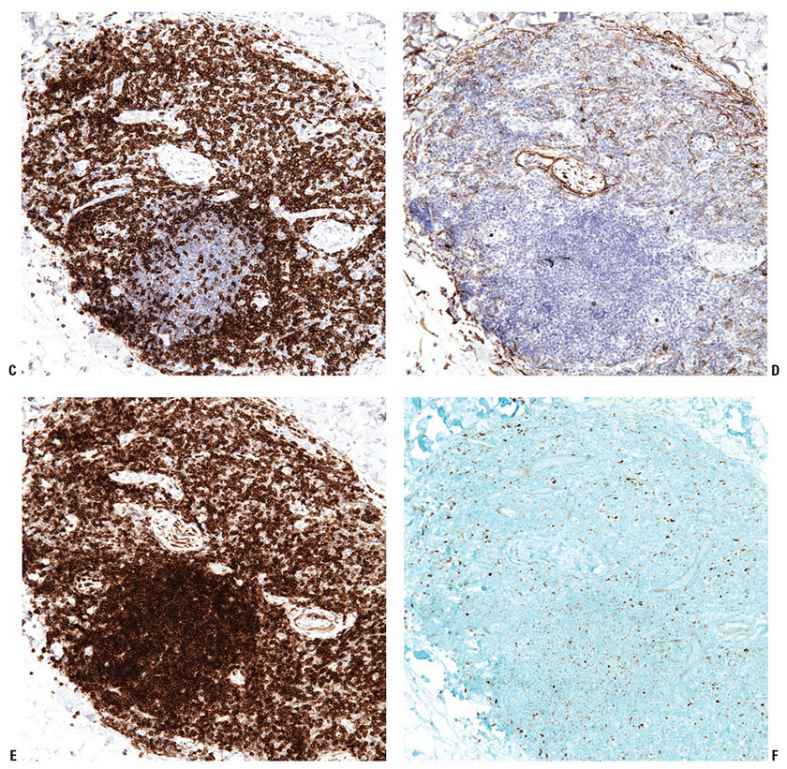
Figure 31-9 Primary cutaneous marginal zone lymphoma. A–F: Immunohistochemistry studies reveal an aggregate of CD20+ B cells (A), surrounded by reactive CD3+ T cells (B). The B cells do not express CD5 (C) or CD10 (D), do express BCL2 (E), and do not express BCL6 (F).
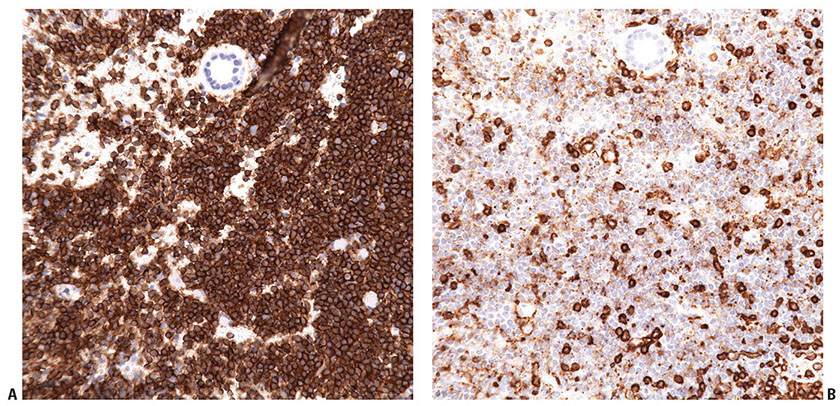
Figure 31-10 Primary cutaneous marginal zone lymphoma. CD43 is often aberrantly expressed in extranodal MALT lymphoma but not in PCMZL. A, B: Immunohistochemistry studies reveal that the CD20+ B cells (A) do not coexpress CD43 (B). The few admixed CD43+ cells seen in this field are reactive small T cells (strong staining) and macrophages (weak staining).
Histogenesis. The cell of origin is speculated to be a post–germinal center marginal zone B cell. Immunoglobulin genes are clonally rearranged in approximately 85% of cases, although it is important to consider that clonal B-cell populations can also be detected in nonneoplastic proliferations (27). In addition, clonal rearrangements of the T-cell receptor (TCR) genes have been found in up to 35% of MALT lymphomas including PCMZL (28). More recently, understanding of the underlying genetic abnormalities in MALT lymphoma has increased. A minority of PCMZL cases demonstrate recurrent chromosomal abnormalities including t(3;14)(p14.1;q32), t(14;18)(q32;q21), and t(11;18)(q21;q21), although none are specific for skin and have also been detected in MALT lymphomas occurring at other sites (29–35). The t(14;18) includes translocations between IGH@ and MALT1 as well as IGH@ and BCL2. FOXP1 on chromosome 3 partners with IGH@ in the documented t(3;14) cases, although this is only seen in about 10% of PCMZL.
Differential Diagnosis. PCMZL may show overlapping features with both neoplastic and nonneoplastic cutaneous B-cell infiltrates (Table 31-3). PCMZL may sometimes lack a monocytoid appearance and thus morphologically mimic secondary involvement by chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) or mantle cell lymphoma (MCL), but these can easily be distinguished by immunophenotyping. The neoplastic cells of PCMZL may sometimes colonize reactive follicles and/or contain increased numbers of centroblasts, raising the possibility of involvement by primary cutaneous follicle center lymphoma (PCFCL), although the expression of BCL6 and CD10 also outside of follicles would favor the neoplastic cells of PCFCL (36). Distinction of PCMZL from a benign reactive infiltrate secondary to injected antigen (arthropod bite) or drug may be very challenging, given the overlapping morphologic features and possibility of clonal immunoglobulin gene rearrangements in benign infiltrates. It may not be possible to distinguish between the two on histologic grounds, in which case correlation with clinical findings is essential. While documentation of clonal plasma cells is a very helpful indicator of PCMZL, it is important to recognize that T-cell lymphomas such as angioimmunoblastic T-cell lymphoma (AITL) may be associated with clonal plasma cell populations (37). This may cause diagnostic difficulty in cases of PCMZL with a rich reactive T-cell infiltrate.
Principles of Management. The disease may spontaneously resolve, or otherwise typically responds well to a variety of therapeutic modalities including radiation therapy, eradication of chronic antigenic stimulus in cases where infectious agents are implicated, and/or treatment with the anti-CD20 monoclonal antibody rituximab.
Primary Cutaneous Follicle Center Lymphoma
PCFCL is a neoplasm derived from follicular center B cells including centrocytes (cleaved follicular center cells) and centroblasts (noncleaved follicular center cells). PCFCL accounts for approximately 10% of cases of cutaneous lymphoma (2,38), and typically follows an indolent clinical course.
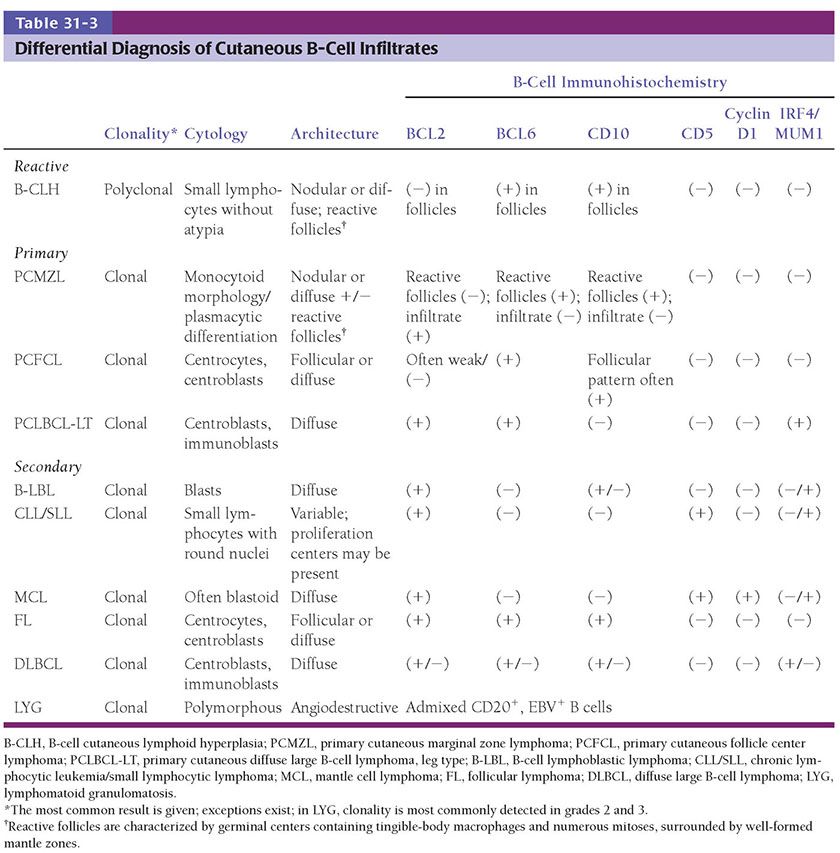
Clinical Summary. PCFCL occurs primarily as one to several red- to plum-colored plaques, nodules, or tumors (Fig. 31-11) often on the head or trunk of adults without sex predilection. PCFCL tends to remain localized in the skin and is often treated with radiation therapy (39). The prognosis of this disease is favorable (5-year survival >95%) despite variations in clinical or morphologic features (localized vs. multifocal disease at presentation; follicular or diffuse growth pattern; number of centroblasts) (2).
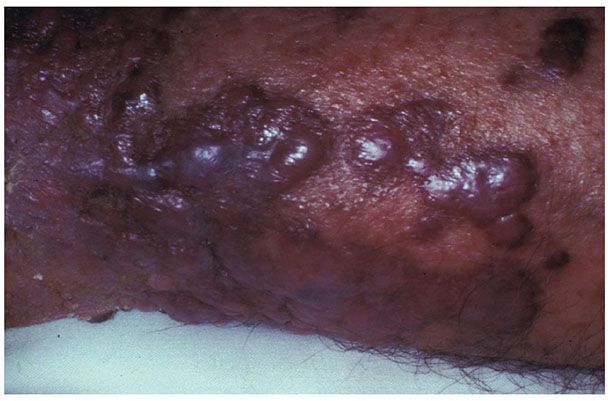
Figure 31-11 Primary cutaneous follicle center lymphoma. Clustered plum-colored nodules covered by thinned, glistening epidermal layer.
Histopathology. The lymphomatous infiltrates occur in the dermis without the involvement of the overlying epidermis. The neoplastic B cells can be arranged in follicles (Fig. 31-12A) or in a diffuse pattern (Fig. 31-12B), or may show a combination of architectural patterns. The neoplastic follicles are depolarized, often devoid of mantle zones and contain monomorphic neoplastic cells without a prominent starry-sky pattern (40). Diffuse areas splay dermal fibers and may demonstrate associated sclerosis. The neoplastic infiltrate consists of medium- to large-sized centrocytes, which demonstrate angulated, folded, or twisted nuclear contours, inconspicuous nucleoli, and scant cytoplasm, as well as large centroblasts, which are transformed lymphocytes with round to oval nuclear contours, vesicular chromatin, and several visible nucleoli. The lesions are often predominantly composed of centrocytes, although an even admixture of centrocytes and centroblasts, or even a predominance of centroblasts may be seen (Fig. 31-12C, D). Occasionally the cells may demonstrate a spindled morphology. Unlike nodal follicular lymphoma (FL), histologic grading of PCFCL is not performed, as the growth pattern and number of blast cells do not appear to have prognostic relevance (41–43). However, tumors showing diffuse, monotonous sheets of centroblasts or immunoblasts are classified as primary cutaneous diffuse large B-cell lymphoma, leg type (PCLBCL-LT) (1).
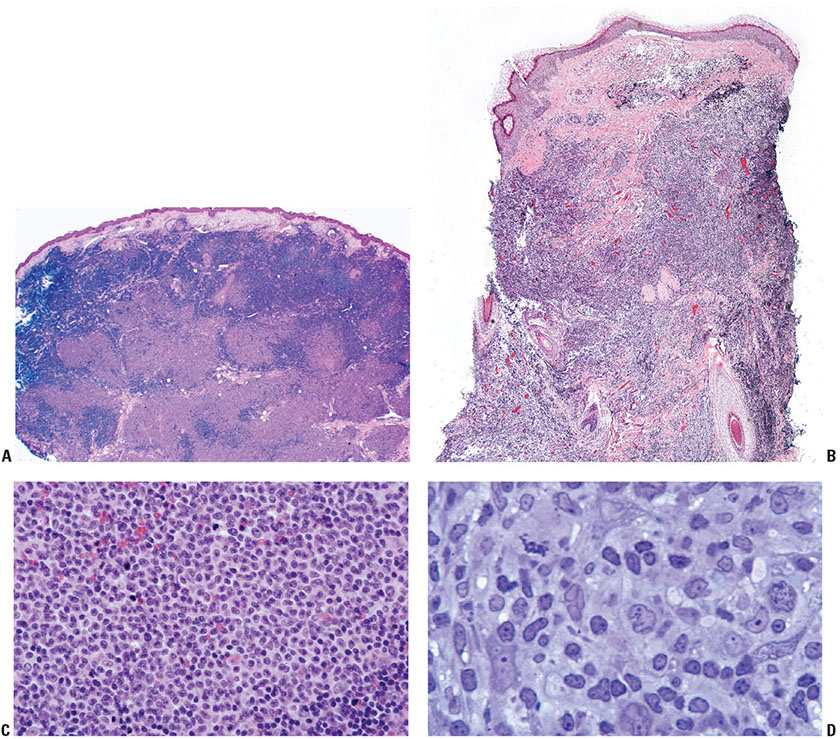
Figure 31-12 Primary cutaneous follicle center lymphoma. A: Scanning magnification showing coalescent nodules forming follicle-like architecture. B: Scanning magnification showing a diffuse infiltrative pattern. C: Higher magnification of malignant centrocytes and centroblasts. D: One-micron, toluidine blue–stained section showing cytologic features of centrocytes and centroblasts.
Tumor cells express the B-cell-associated markers CD20, CD19, CD22, and CD79a, but are usually negative for immunoglobulin. BCL6 expression is a constant feature; CD10 expression is more common in cases with a follicular architecture; and interferon regulatory factor-4/multiple myeloma-1 (IRF4/MUM1) staining is typically negative (Fig. 31-13A–F). BCL2 is frequently weak or negative in PCFCL (Fig. 31-13G), although strong, distinct expression has been described in a subset of cases, particularly those with follicular architecture (44–46). Markers of follicular dendritic cells (such as CD21 or CD35) can be used to detect underlying, associated follicular dendritic cell meshworks in cases with at least partial follicular architecture (Fig. 31-13H). In cases with a diffuse pattern, these stains may highlight few residual follicular dendritic cells or may be completely negative (Fig. 31-14A–C). The Ki67 proliferation index may be high, particularly in diffuse cases composed of large centrocytes (47).
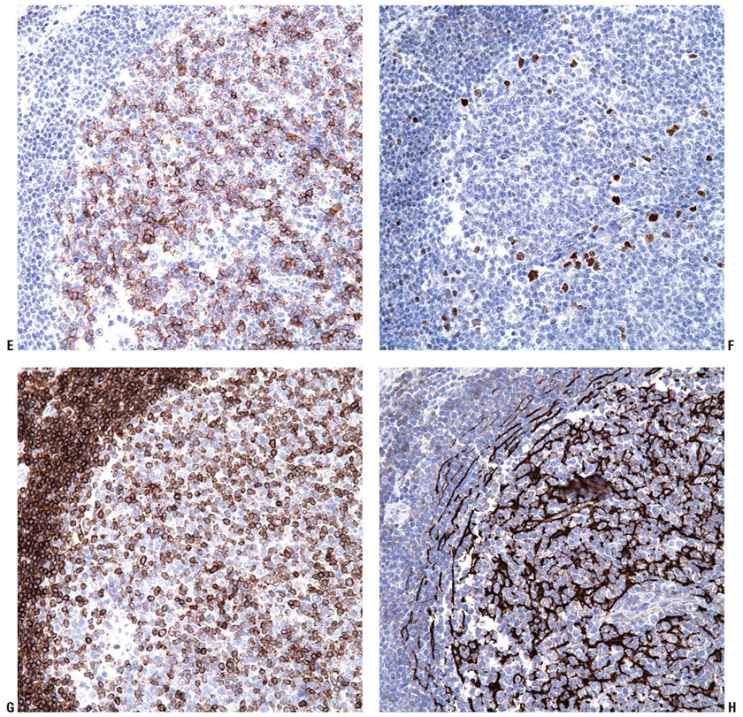
Figure 31-13 Primary cutaneous follicle center lymphoma. A–C: Immunohistochemistry studies performed on a neoplastic follicle (A) reveal that the follicles are composed of CD20+ B cells (B) and show few infiltrating reactive CD3+ T cells (C). D–F: The B cells commonly coexpress BCL6 (D), CD10 (E), and are negative for IRF4/MUM1 (F). G: Weak, heterogeneous expression of BCL2 may be seen in the B-cell follicles. H: CD21 will highlight the underlying follicular dendritic cell meshworks.
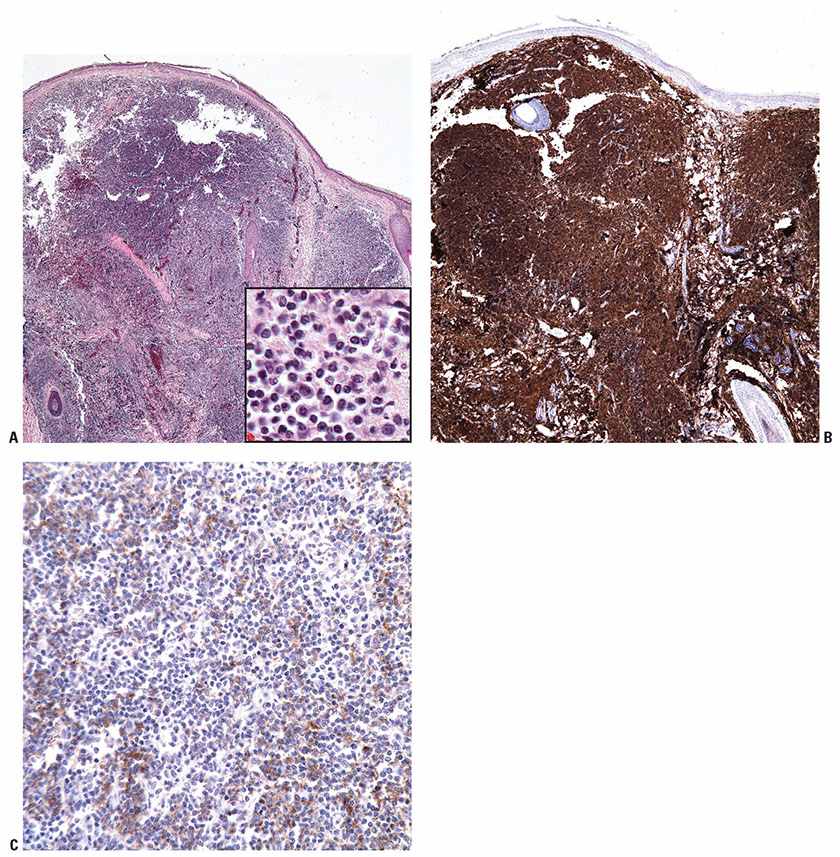
Figure 31-14 Primary cutaneous follicle center lymphoma (PCFCL). A: Diffuse pattern of PCFCL seen at scanning magnification, with inset showing small-sized cells at higher power. B: CD20 marks the diffuse B-cell infiltrate. C: In contrast to the prior figure, staining with CD21 reveals weak, nonspecific expression and does not highlight an underlying follicular dendritic cell meshwork.
Histogenesis. Germinal center–derived B cells are believed to represent the cell of origin of PCFCL. The t(14;18)(q32;q21) translocation is detected only in a subset of cases, apparently more consistently with fluorescence in situ hybridization (FISH)-based analysis compared to polymerase chain reaction (PCR) (46,48,49). Interestingly, BCL2 expression by immunohistochemistry can be detected in cases without an underlying t(14;18) (46,50). Overall, these findings implicate the role of the IGH-BCL2 translocation as well as other genetic alterations in the pathogenesis of PCFCL. The presence or absence of BCL2 expression and/or t(14;18) does not impact prognosis. Clonal rearrangement of immunoglobulin genes is often detected.
Differential Diagnosis. PCFCL may show overlapping features with both neoplastic and nonneoplastic cutaneous B-cell infiltrates (Table 31-3). Distinction from B-cell cutaneous lymphoid hyperplasia (B-CLH), which may show similar clinical features, is necessary. The typical features of the reactive follicles in B-CLH (well-formed mantle zones, polarized germinal centers with tingible-body macrophages (macrophages containing particulate basophilic debris within their cytoplasm), and numerous mitotic figures) are morphologically distinct from the monomorphous follicles of PCFCL. In general, clonality studies will reveal a polyclonal B-cell population in B-CLH and a clonal B-cell population in PCFCL, although exceptions do exist, and therefore incorporation of morphologic and immunophenotypic findings is also needed. In addition, PCFCL must be differentiated from secondary cutaneous involvement by nodal-type FL. Although there are no absolute morphologic or immunophenotypic features that can reliably distinguish the two entities, strong immunoreactivity for both BCL2 and CD10 and/or detection of t(14;18) raises the possibility of systemic FL. In any case lacking staging information, a differential diagnosis should be provided. Another potential diagnostic difficulty is the distinction between PCFCL and PCMZL demonstrating follicular colonization or associated reactive follicles. Assessing the immunophenotype of the interfollicular cells may help; PCFCL displays a BCL2−/+/CD5−/CD10+/−/BCL6+ phenotype, while PCMZL cells are BCL2+/CD5−/CD10−/BCL6− (51). Finally, morphologic distinction between PCFCL with a purely diffuse pattern from PCLBCL-LT is difficult, especially in cases where centroblasts predominate in the PCFCL infiltrate. Immunohistochemical profiling may be useful in this scenario, although it is essential to recognize that exceptions do exist. Both entities express BCL6, while CD10 is often negative in the latter. In addition, PCFCL may lack strong BCL2 and often lacks IRF4/MUM1 expression and forkhead box P1 (FoxP1) expression, all of which are strongly positive in PCLBCL-LT (2,52,53). Additionally, frequent cytoplasmic expression of IgM (and less commonly also IgD) is detected in PCLBCL-LT, while detection of either of these heavy chains is uncommon in PCFCL (54,55).
Principles of Management. PCFCL is typically an indolent disease, and solitary or localized lesions may be treated with radiation therapy or excision. Multifocal lesions may be treated with radiation, topical agents, or steroid injections. Systemic chemotherapy is rarely indicated. Cutaneous relapse is observed in approximately 30% of cases, but does not portend a poorer prognosis (39). Of note, presentation on the leg does appear to have a less favorable prognosis and warrants close clinical management (52,56).
Primary Cutaneous Diffuse Large B-Cell Lymphoma, Leg Type
PCLBCL-LT is an aggressive primary cutaneous large B-cell lymphoma characterized by a diffuse, cutaneous infiltrate of immunoblasts or centroblasts in a sparse reactive background and strong immunoreactivity for IRF4/MUM1 and BCL2 (57,58).
Clinical Summary. PCLBCL-LT most commonly presents as solitary or multiple localized reddish-brown tumors. Although the disease typically presents on the leg, lesions with a similar morphology and phenotype can arise at any cutaneous site. Affected patients are usually elderly, with a female predominance. The overall prognosis is intermediate to poor, with a 5-year survival of 55% and a tendency to disseminate to extracutaneous sites. Ulceration, primary manifestation on the leg, or multiple lesions at presentation are associated with adverse prognosis (53,59,60).
Histopathology. PCLBCL-LT manifests as a dense, monotonous infiltrate of predominantly medium- to large-sized cells with round nuclear contours and prominent nucleoli (immunoblasts or centroblasts) (Fig. 31-15A, B). Frequent mitoses are seen, and reactive small lymphocytes are sparse. The infiltrate diffusely involves the dermis, obliterating the adnexa, and often extends into the underlying subcutaneous tissue. Focal epidermotropism with Pautrier-like microabscesses may be seen. The neoplastic cells strongly express IRF4/MUM1 and BCL2 but not CD10 (Fig. 31-16A–E). Most cases also express BCL6, a marker for B-cell germinal center differentiation, as well as IgM (Fig. 31-17A, B) (54,55). Strong FoxP1 expression is also common. The Ki67 proliferation index is usually high (approximately 70%) (52). CD30 expression may be detected, but this does not change the classification if the case otherwise meets morphologic and immunophenotypic criteria for this entity (61).

Figure 31-15 Primary cutaneous diffuse large B-cell lymphoma, leg type. A: Scanning magnification view shows a diffuse lymphoid infiltrate encompassing the entire dermis but separated from the overlying epidermis by a distinct Grenz zone. B: The neoplastic infiltrate is composed of large-sized cells with open chromatin and distinct nucleoli. Mitotic figures are abundant.
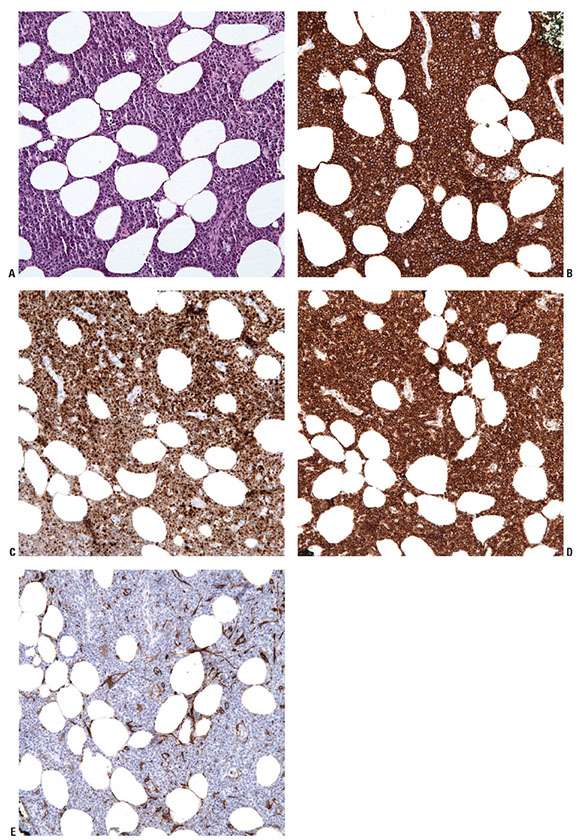
Figure 31-16 Primary cutaneous diffuse large B-cell lymphoma, leg type. A–E: Immunohistochemistry studies reveal that PCLBCL-LT, here shown in the subcutis (A), is composed of CD20+ B cells (B) that strongly coexpress IRF4/MUM1 (C) and BCL2 (D), and do not express CD10 (E).
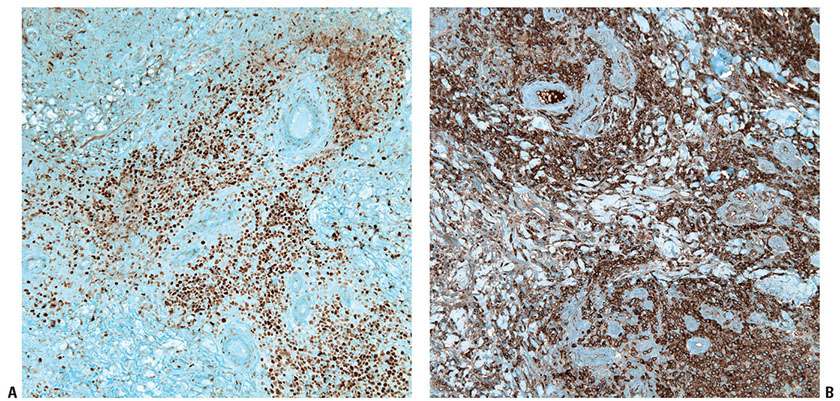
Figure 31-17 Primary cutaneous diffuse large B-cell lymphoma, leg type. A, B: Immunohistochemistry studies demonstrate strong expression of BCL6 (A) and IgM (B) in PCLBCL-LT.
Histogenesis. PCLBCL-LT has a B-cell differentiation profile of post–germinal center (activated) B cells (62). Molecular profiling demonstrates a signature in PCLBCL-LT distinct from other types of cutaneous B-cell lymphomas (62–64). The t(14;18) translocation involving IGH@ and BCL2 is not found in this entity. Unlike other cutaneous B-cell lymphomas that appear to be genetically unrelated to their nodal-based counterparts, cytogenetic studies of PCLBCL-LT have demonstrated frequent translocations similar to those in systemic DLBCL involving IGH@, MYC, and BCL6 loci (31), as well as the presence of the MYD88 oncogenic L265P mutation (65,66). Approximately 75% of cases exhibit inactivation of 9p21.3/CDKN2A via deletion or promoter hypermethylation, which is a poor prognostic indicator (64,67,68).
Differential Diagnosis. The differential diagnosis includes PCFCL, diffuse type or systemic DLBCL secondarily involving the skin (Table 31-3). Cytomorphology may provide clues to the diagnosis (cleaved medium-sized centrocytes with inconspicuous nucleoli in PCFCL vs. large, round centroblasts with prominent nucleoli in PCLBCL-LT). Occasionally, cases of PCFCL may exhibit an admixture of centrocytes and centroblasts or even predominance of centroblasts. In the latter scenarios, immunohistochemical profiling is paramount for diagnosis. In contrast to PCLBCL-LT, which strongly expresses BCL2, IRF4/MUM1, and IgM, PCFCL diffuse type is typically negative for these markers. Epstein–Barr virus (EBV)+ DLBCL of the elderly may involve the skin but is distinguished by positive in situ hybridization for EBV-encoded RNA (EBER), which is negative in PCLBCL-LT.
Principles of Management. Radiotherapy, systemic chemotherapy, and/or rituximab administration are mainstays of treatment. Regimens may need to be tailored to the underlying fitness of the patient given the frequent occurrence in elderly individuals.
Mature T-/Natural Killer–Cell Lineage
Mycosis Fungoides
MF is the prototype of cutaneous T-cell lymphoma (CTCL) that typically begins as slowly progressive dermatitis-like patches and plaques that may evolve to nodules or tumors. Eventual systemic dissemination will occur in a subset of patients (69). The patch/plaque stage of the disease is the result of intraepidermal and superficial dermal infiltration by small- to medium-sized neoplastic T cells with characteristically cerebriform nuclear contours. More advanced stages develop as a consequence of dermal or extracutaneous involvement by cytologically more atypical T cells. Diagnosis at early stages may be challenging solely on histologic grounds given the morphologic overlap with other (predominantly reactive) conditions, and requires careful clinicopathologic correlation. Application of a validated clinicopathologic algorithm developed by the International Society for Cutaneous Lymphoma may be helpful in suspected early MF (70,71) (Table 31-4). Multiple biopsies may be necessary to establish the diagnosis. TCR gene rearrangements may or may not be present in MF, and reactive conditions may also show clonal TCR gene rearrangements, limiting the usefulness of this ancillary study in early-stage disease. However, demonstration of identical clones at different anatomic sites or over time will support a diagnosis of a neoplastic process.
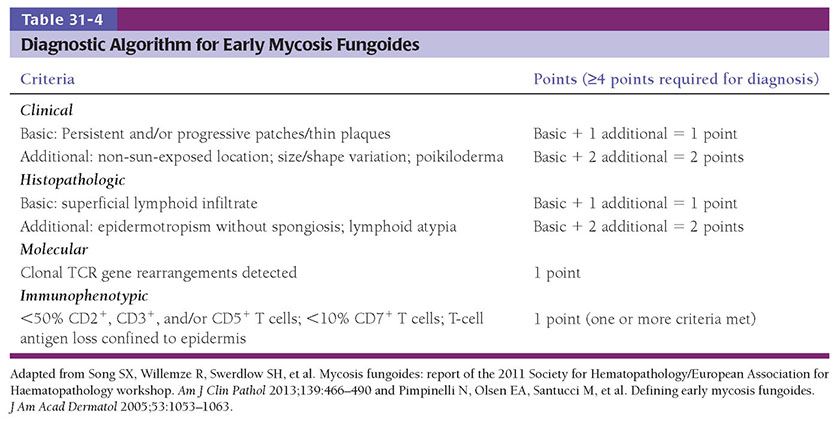
Clinical Summary. MF is relatively rare, accounting for less than 1% of non-Hodgkin lymphomas. However, of lymphomas that arise primarily in skin, it is the most common, accounting for approximately 44% of cutaneous lymphomas (72). It is a disease predominantly of adults, with a male-to-female ratio of 2:1, although children are occasionally affected. MF has a plethora of clinicopathologic manifestations. Typical lesions initially present as large, erythematous scaling patches and plaques that may show arcuate or polycyclic configurations and are generally resistant to anti-inflammatory therapy (Fig. 31-18A). A number of distinct variants are described in the sections below. As the disease advances, larger plaques and nodules and even frank tumors will develop, often with associated ulceration (Fig. 31-18B), although patches and plaques are often still present at this stage. If extracutaneous spread occurs, the lymph nodes, spleen, liver, and lungs are often involved, in addition to the peripheral blood. Lymph node enlargement at such advanced stages must be differentiated from nodal hyperplasia resulting from chronic drainage of involved sites (dermatopathic lymphadenopathy). An erythrodermic manifestation may occur, but is uncommon.
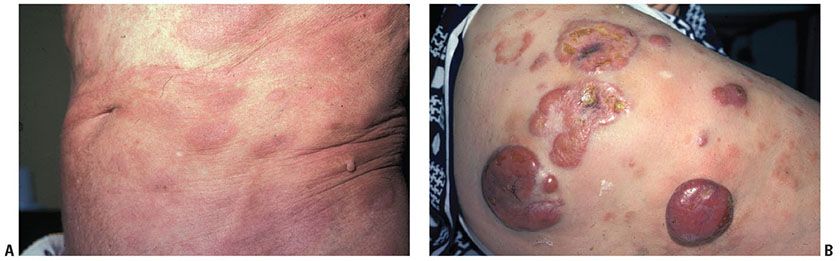
Figure 31-18 Mycosis fungoides: clinical features. A: Erythematous scaling patches and plaques of early disease. B: Plaques and nodules of advanced disease.
Clinical staging follows criteria uniquely developed for MF and also used for Sézary syndrome (SS), and incorporates evidence of histologic, immunophenotypic, and/or molecular involvement of cutaneous, nodal, and visceral sites as well as peripheral blood (73). Patients do not inevitably progress through the disease stages. For example, in a recent large study, 71% of patients presented with early-stage disease and disease progression occurred in 34% of patients (74). For those that do progress, higher stage is directly correlated to decreased survival. Patients with stage IA disease (limited patches, papules, and/or plaques covering <10% of the skin surface with or without low blood tumor burden) have an equivalent life expectancy to healthy age-matched controls, those with stage IB (≥10% of the skin surface) have a median survival of 10 to 12 years, those with tumors or associated erythroderma have a median survival of 4 to 5 years, and those with extracutaneous visceral involvement have a median survival of 1 to 2 years (69,72,74–80). Poor prognostic indicators include elevated lactate dehydrogenase levels, male gender and advanced age (74), and black race (81).
Histopathology. Cytologically, the neoplastic T cells in MF are small- to medium-sized lymphocytes that characteristically contain nuclei with dense heterochromatin and elaborately indented (cerebriform) nuclear contours (Fig. 31-19A, B). These cells may be difficult to differentiate from cytokine-activated T cells, although one helpful feature is the tendency for epidermotropic neoplastic T cells to predominantly or exclusively show cerebriform nuclear features and clear perinuclear halos.
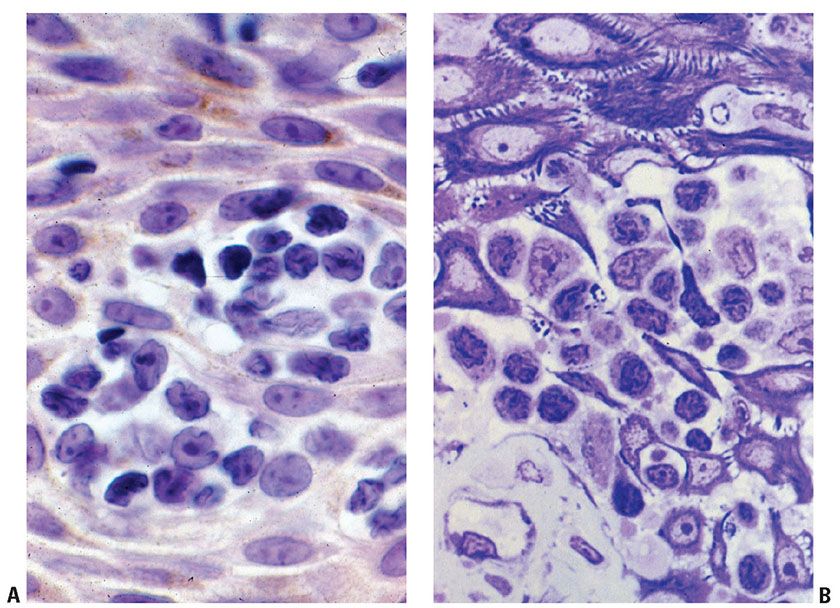
Figure 31-19 Mycosis fungoides: cytology. A: Oil immersion conventional microscopy of atypical lymphocytes within the epidermal layer. B: One-micron, toluidine blue–stained section of epidermotropic malignant lymphocytes showing characteristically infolded nuclear contours.
The clinical manifestations of MF (patch, plaque, or tumor stage) roughly correlate with the microscopic appearance of the lesions. Early lesions of MF show a patchy bandlike lymphocytic infiltrate within the papillary dermis, often associated with coarse fibrosis (Fig. 31-20A, B). Often, only a few of the atypical T cells are present in the dermis and are often obscured by a background of many small (reactive) T cells as well as admixed eosinophils, plasma cells, and histiocytes. This can make the diagnosis of early-stage MF very challenging, and indeed in some instances definitive diagnosis is not possible. Helpful clues for the diagnosis of MF include the linear accumulation of atypical lymphocytes along the basement membrane zone of the epidermis (a lentiginous pattern frequently described as tagging) (Fig. 31-21A, B) or a single-cell pagetoid infiltrate of atypical lymphocytes into the epidermis (epidermotropism) (Fig. 31-21C). Lymphoid epidermotropism in MF, unlike lymphoid exocytosis in dermatitis, is generally not associated with significant epidermal spongiosis or evidence of cytotoxicity (basal cell layer vacuolization or apoptosis). Rather, the epithelium appears “passive,” resulting in an accumulation of atypical lymphocytes within lacunar spaces that separate seemingly permissive keratinocytes. Exceptions, of course, do exist, and infrequently marked spongiosis or an interface dermatitis may be seen in MF.
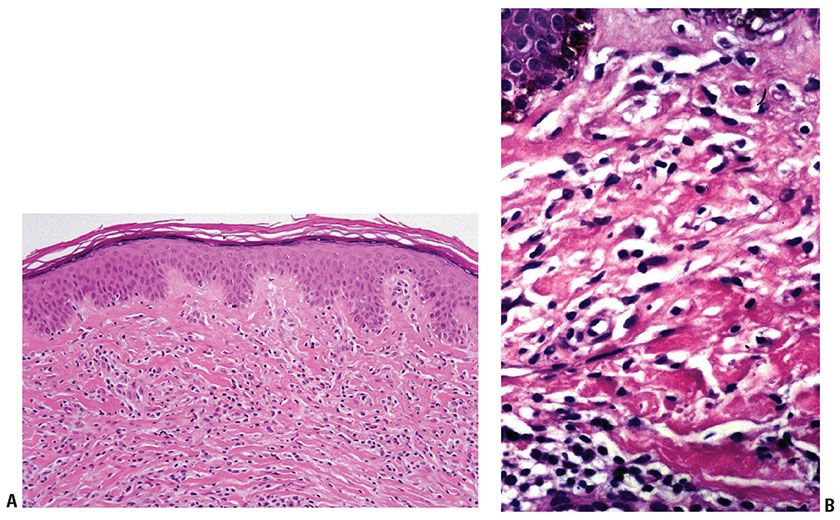
Figure 31-20 Mycosis fungoides: histologic features of early disease. A, B: Papillary dermal interstitial lymphoid infiltration associated with coarse fibrosis.
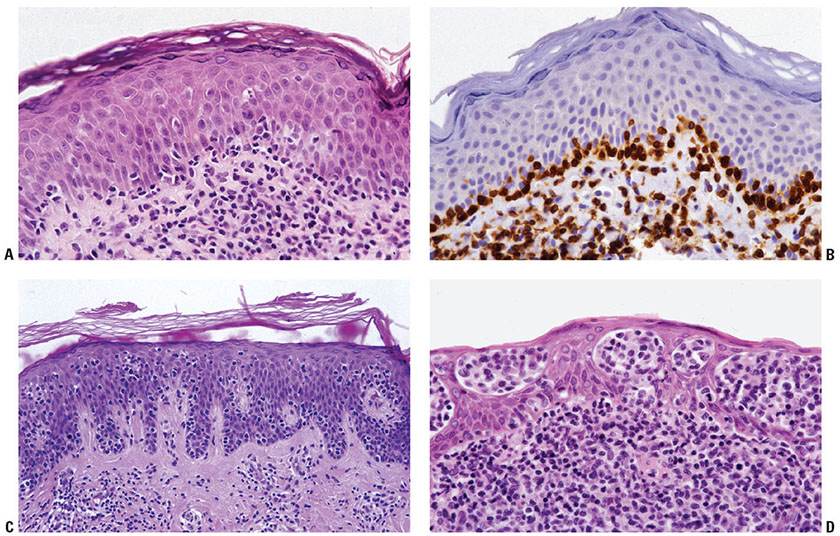
Figure 31-21 Mycosis fungoides: architectural patterns of epidermotropism. A: Papillary dermal interstitial infiltrate associated with linear aggregation of neoplastic lymphocytes along the dermal–epidermal junction. B: Adjacent section stained for CD4 emphasizing this linear pattern of early epidermotropism. C: Early plaque-stage disease with single-cell epidermotropism by atypical lymphocytes into a “passive” epidermal layer. D: Plaque-stage disease characterized by prominent clusters of atypical lymphocytes within the epidermis (Pautrier microabscesses).
The presence of clustered epidermotropic atypical lymphocytes, known as Pautrier microabscesses (Fig. 31-21D), is uncommonly observed in early patch-stage MF but frequently encountered in plaque-stage MF. These intraepithelial aggregates of neoplastic T cells must be differentiated from similar aggregates of larger Langerhans cells, which also demonstrate irregular, frequently indented, nuclear profiles; these so-called Langerhans cell microgranulomas are commonly associated with dermatitis (Table 31-5) (14). In plaque-stage disease, the bandlike dermal infiltrate is denser than in patch stage and the atypical T cells are abundant. An interstitial pattern may also be seen.
In more advanced stages, there is often loss of epidermotropism. The dermal infiltrate is characterized by a nodular to diffuse interstitial population of cytologically atypical T cells, which may extend into the subcutaneous tissues (Fig. 31-22A). These cells may retain the small/medium size of earlier stages (Fig. 31-22B), or may demonstrate transformation to large, highly atypical cells (large-cell transformation or LCT) (Fig. 31-22C). LCT is defined as large-sized cells with prominent nucleoli composing 25% or more of the infiltrate or forming microscopic nodules (82), and is seen in greater than 50% of tumor-stage lesions (83). LCT is associated with the development of an aggressive biologic course (84,85), although some suggest that a subset of cases may show more indolent behavior (86).
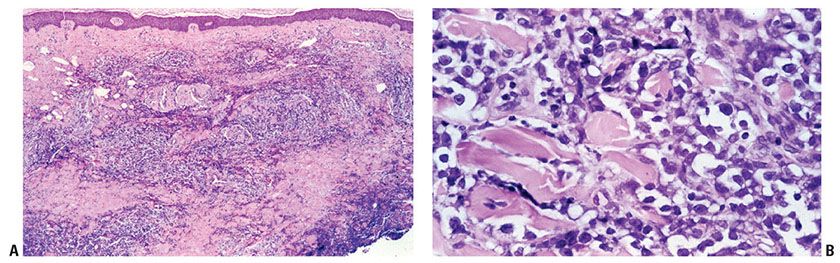
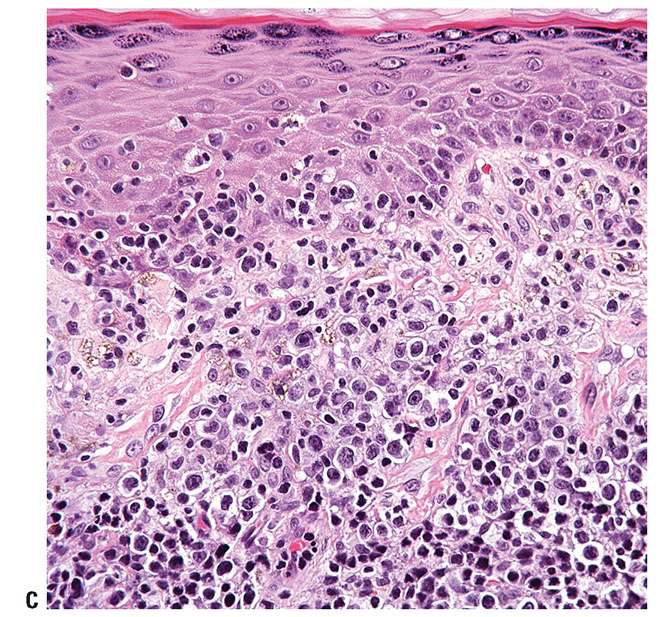
Figure 31-22 Mycosis fungoides: advanced stage. A: There is a nodular to diffuse dermal infiltrate with relative sparing of the superficial dermis and epidermis. B: In some cases, atypical lymphocytes retain the small/medium size of earlier stages. C: In other cases, transformation to large, highly atypical cells (large-cell transformation) occurs.
MF cells are most commonly αβ helper T cells with an immunophenotype of CD3+, CD4+, CD5+, and CD8− (Fig. 31-23A–F), although occasionally a CD8+ αβ immunophenotype is observed (87). Lesions at all stages may show absence of CD7, although this finding in itself does not reliably permit differentiation of MF cells from reactive T cells (87). Loss of CD3, CD2, and/or CD5 expression in lesional T cells provides support for a neoplastic infiltrate, although this helpful sign is not common at early stages. Particularly in early stages, careful evaluation of marker expression (or lack thereof) in the intraepidermal cells is necessary given the potentially confounding reactive T-cell infiltrate in the dermis. Cytotoxic markers (T-cell intracellular antigen [TIA-1], perforin, granzyme B) are negative in patch/plaque-stage disease but subset expression may be detected at tumor stage (88). The skin-homing antigen cutaneous lymphocyte-associated antigen (CLA) is expressed in most cases. Although occasional CD30+ cells may be identified in the dermal infiltrates, this epitope is not expressed by most cells comprising the malignant clone. However, in advanced tumor-stage disease, especially with LCT, the cells may acquire CD30 expression (Fig. 31-24A, B). It is important to recognize that CD30 expression is not a prerequisite for the diagnosis of LCT, as not all cases of LCT demonstrate CD30 expression.
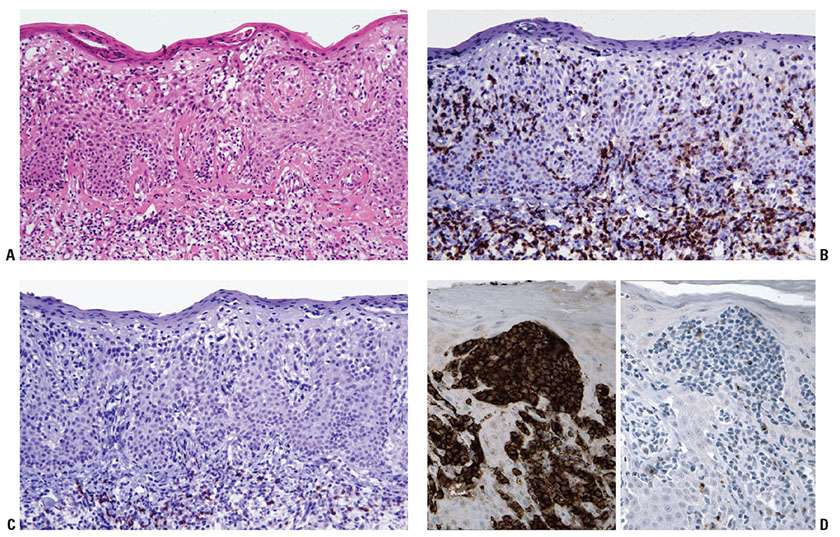
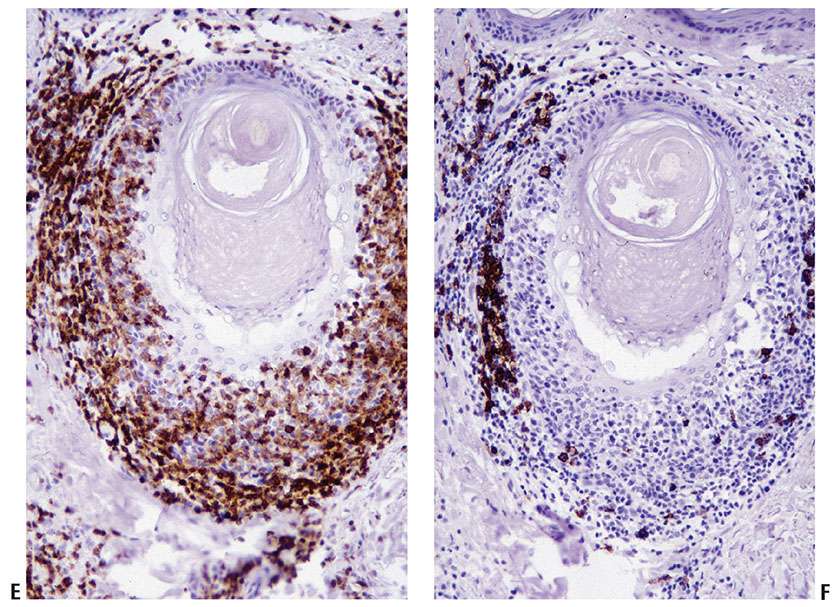
Figure 31-23 Immunophenotypic features of mycosis fungoides. A–C: Serial sections through a plaque-stage lesion with marked epidermotropism (A) reveal that most of the epidermotropic cells (in particular, those displaying nuclear atypia) express CD4 (B) but not CD8 (C). D: In addition, the epidermotropic CD4+ malignant T cells (left panel) may exhibit aberrant loss of other T-cell markers, such as CD7 (right panel). E, F: Similar findings are observed for folliculotropic T cells, which express CD4 (E) but not CD8 (F).
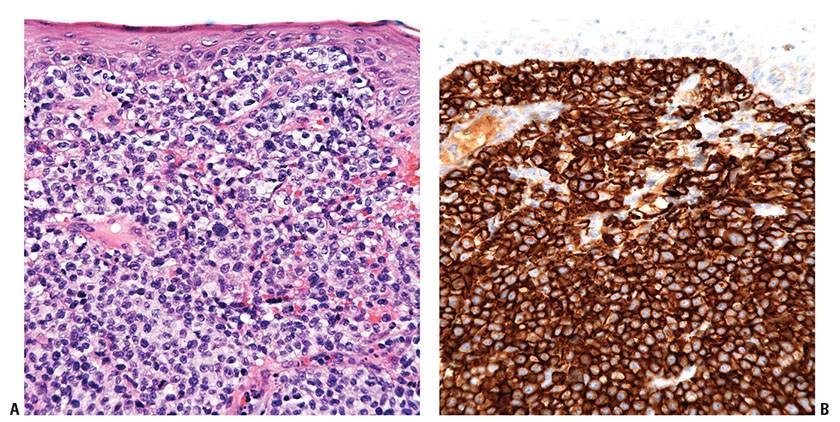
Figure 31-24 Mycosis fungoides with large-cell transformation. A, B: Immunohistochemistry studies reveal that the diffuse infiltrate of large, neoplastic cells (A) can acquire homogenous, strong expression of CD30 (B).
Apart from the classic MF described above, there are a number of clinicopathologic variants and subtypes of MF. The clinically significant variants as recognized by the 2008 WHO classification scheme (1) are the following:
Folliculotropic mycosis fungoides. A diagnosis of folliculotropic MF predicts poor survival and increased risk of disease progression compared to typical MF (74,76). Folliculotropic MF is characterized by infiltration of hair follicles by atypical T cells with minimal to absent epidermotropism, and thus the diagnosis may be overlooked when biopsies are too superficial. Often, but not always, there is associated follicular mucinosis secondary to intracellular mucin production by keratinocytes (89). These cells eventually degenerate, culminating in “alopecia mucinosa” (Fig. 31-25A–C). Other features include marked reactive lymphoid infiltrates, granulomatous reaction, abundant eosinophils or plasma cells, or cyst formation (90,91). Clinically, in addition to patchy alopecia, folliculotropic MF may manifest as follicular papules and plaques or comedones, and can be associated with intense pruritus. This form of the disease may occur anywhere, although the skin of the face and the scalp as well as sun-exposed extremities are often involved. The deeper localization of the neoplastic infiltrate in folliculotropic MF may render reduced response to skin-targeted therapies. Although not formally included in this category, eccrine involvement by MF (syringotropic MF) has rarely been described, with or without follicular lesions (91). Infiltration of the eccrine coil may be associated with syringometaplasia but not mucinosis (Fig. 31-25D).
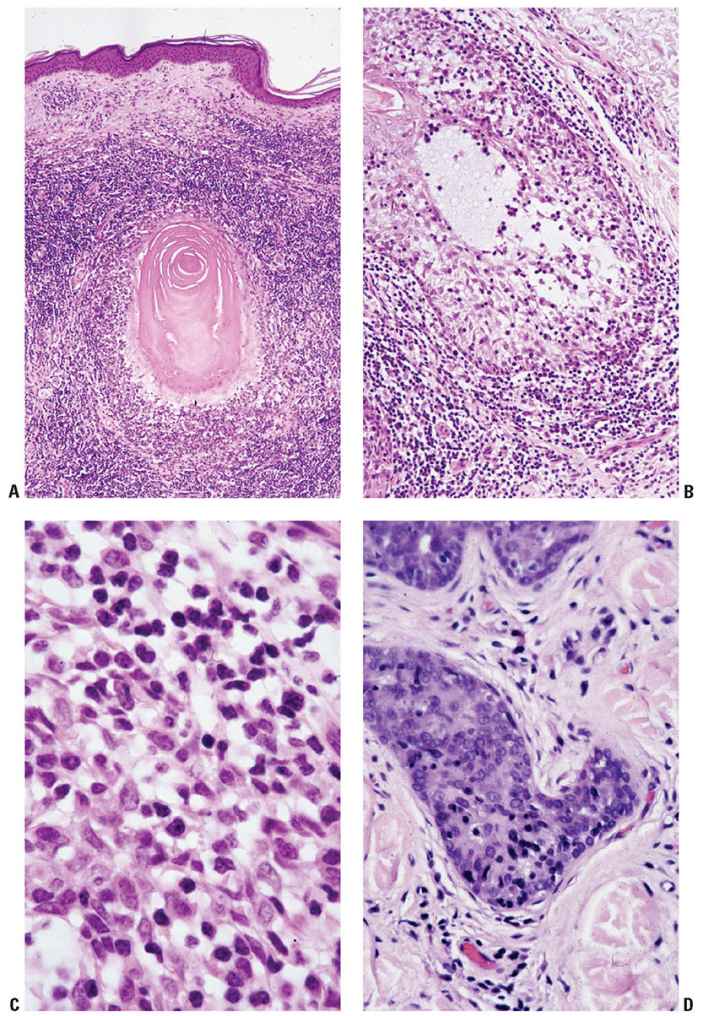
Figure 31-25 Adnexotropic mycosis fungoides. A: Infiltration is associated with architectural distortion and destruction of follicular structures. B: Follicular mucinosis characterized by pools of pale blue–staining mucin. C: Atypical T cells in a mucinous background. D: Eccrine involvement by atypical lymphocytes, associated with syringosquamous metaplasia.
Localized pagetoid reticulosis (Woringer–Kolopp disease). Localized pagetoid reticulosis (Woringer–Kolopp disease) is a rare form of MF characterized by epidermal infiltration of medium- to large-sized neoplastic T cells with abundant pale cytoplasm, often in the form of a solitary acral lesion where associated epidermal acanthosis and hyperkeratosis results in the clinical impression of a squamous proliferation such as a verruca or eczema. The pagetoid growth pattern of the neoplastic T cells may be attributed to their strong expression of adhesion molecules, in particular CLA and αEβ7, which facilitate interactions with endothelial cells and keratinocytes, respectively (92). The neoplastic T cells are either CD4+ or CD8+ and CD30 expression may be seen (93). Histopathologically, because of the pronounced epidermotropism, pagetoid reticulosis may mimic superficial spreading melanoma, pagetoid squamous carcinoma in situ, or extramammary Paget disease. However, diagnosis can be readily achieved on the basis of cytomorphologic and immunophenotypic features. The outcome of localized pagetoid reticulosis is very good. Cases previously described as generalized pagetoid reticulosis (Ketron–Goodman disease) are distinct from this entity and represent, for the most part, examples of aggressive CD8+ T-cell lymphoma.
Granulomatous slack skin (GSS). This rare subtype is characterized by the presence of elastolytic granulomas that develop in association with a dermal lymphomatous component, which is typically dense and extends deep into the subcutis (Fig. 31-26A, B). Clinically, zones of skin with increased folding and decreased elasticity gradually develop, with preferential involvement of intertriginous sites, as a result of loss of elastic fibers. Histologically, atypical T-cell infiltrates are intimately associated with noncaseating dermal granulomas formed by histiocytes and multinucleated giant cells (Fig. 31-26C) that may contain ingested elastic fibers (elastophagocytosis) or engulfed lymphocytes (lymphophagocytosis) (94). In established lesions, there is marked reduction of elastic fibers (Fig. 31-26D). The disease demonstrates an indolent clinical course, and may arise in association with preexisting MF or CHL.
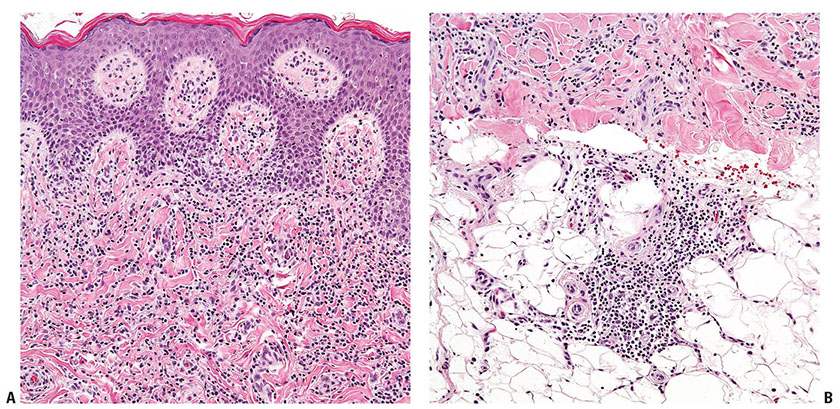
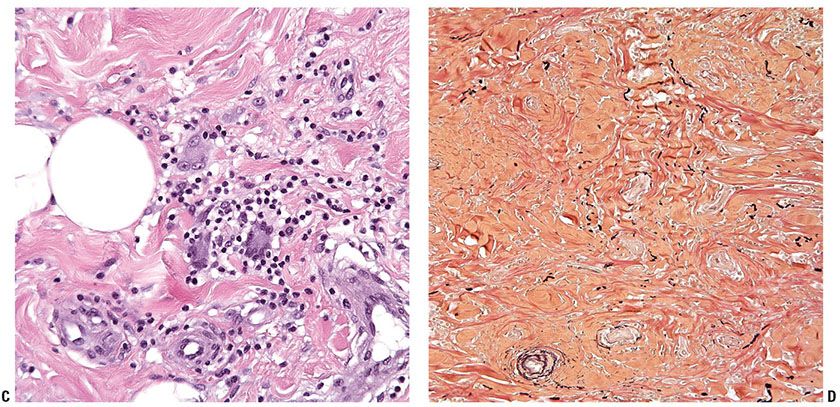
Figure 31-26 Granulomatous slack skin. A, B: The dermal lymphocytic infiltrate (A) extends into the underlying subcutis (B). C: Large, multinucleated giant cells are admixed among the lymphocytes and histiocytes. D: An elastin stain reveals a marked reduction of elastic fibers.
In addition to those listed above, other variants of MF have been described. Despite the presence of characteristic features that permit histologic segregation into distinct categories, these variants share a similar clinical course and prognosis with classical MF and thus do not have prognostic significance. Nonetheless, because of their clinical and histopathologic similarity to reactive dermatoses, diagnosis of these variants can be challenging and familiarity with their clinical and histologic manifestations is necessary (95). A number of these variants are described below.
Acanthosis nigricans–like (vegetating/papillomatous) mycosis fungoides. As the name implies, the lesions often resemble acanthosis nigricans or seborrheic keratosis clinically and have a predilection for flexural sites (axillae and groin), neck, nipple, and areolae (96,97). Microscopically, the lesions have combined features of seborrheic keratosis and typical MF (e.g., a bandlike atypical lymphoid infiltrate with epidermotropism, elongated interconnected rete ridges, and pseudo–horn cysts).
Bullous (vesicular) mycosis fungoides. Bullous MF manifests clinically as blisters that may develop on normal skin or within typical plaques and tumors of MF (98–101). This variant may be of significant clinical relevance in terms of prognosis, as in one study, almost 50% of patients died within a year following the onset of blisters (99). In addition to common histologic features of classical MF, there is associated clefting, which may occur at different planes (subcorneal, intraepidermal, and subepidermal). Although the precise mechanism of the blister formation is not clear, it has been proposed that the subcorneal and intraepidermal separation may result from confluence of Pautrier microabscesses, and the subepidermal blister may be secondary to reduced keratinocyte adherence to the basement membrane in response to cytokines produced by the lymphoma cells (102). Unlike immunobullous disorders, immunofluorescence studies are consistently negative in bullous MF.
Granulomatous mycosis fungoides. Clinically, the lesions present as papules, plaques, and tumors that are not distinctive from classical cases (103). Histopathologically, granulomatous MF is characterized by the presence of typical MF as well as variable granulomatous inflammation, which may take the form of interstitial, tuberculoid/sarcoidal, or palisading granulomata reminiscent of granuloma annulare and/or necrobiosis lipoidica (104–106). Multinucleated giant cells, usually of the foreign-body type (although Langerhans and Touton giant cells have been described as well) (Fig. 31-27A–C) with or without focal elastophagocytosis, may be seen (103,107,108). Special stains for microorganisms are useful in excluding concurrent infection in a patient with classical MF. Granulomatous MF can be differentiated from GSS clinically by the absence of bulky skin lesions. Lymphophagocytosis and elastophagocytosis are less common than in GSS (109).
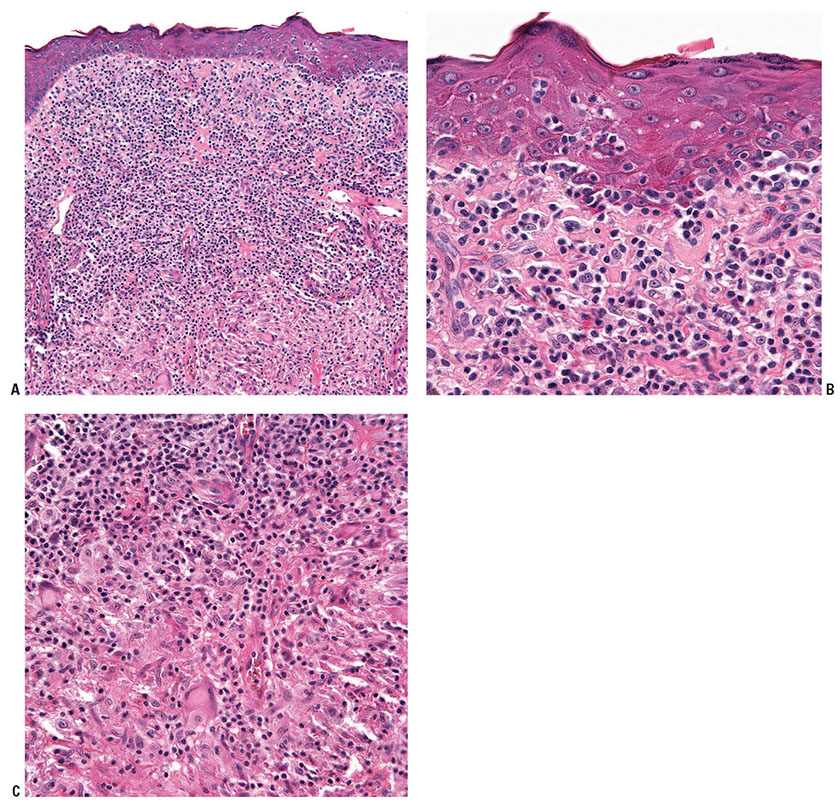
Figure 31-27 Granulomatous mycosis fungoides. A–C: High-powered views of a biopsy from a patient with this variant (A) reveal epidermotropism of the atypical T cells (B) as well as multifocal giant cells admixed within the infiltrate (C).
Hyperpigmented mycosis fungoides. This is a rare variant characterized by diffuse macular hyperpigmentation (110). Histopathologically, there is often marked elongation of the rete ridges with basal hyperpigmentation and dermal melanophages superimposed on features of classical MF. Ultrastructural studies have revealed giant melanin granules in the tumor cells, keratinocytes, Langerhans cells, and macrophages.
Hypopigmented mycosis fungoides. This variant is predominantly seen in young patients with dark skin (111,112). The lesions appear as asymptomatic or slightly pruritic, hypopigmented patches that may or may not be accompanied by typical patches and plaques of classical MF. The underlying mechanisms leading to hypopigmentation have been a subject of controversy. Some studies have found degenerative changes in melanocytes secondary to nonspecific cellular injuries, while others demonstrated impaired melanosome transfer to keratinocytes. Apart from prominent pigment incontinence (Fig. 31-28), hypopigmented MF is indistinguishable from classical MF histopathologically. Although a frequent CD8+ phenotype has been described in the former, the two variants share similar clinical course and prognosis (113). Distinction from vitiligo can be challenging and may only be resolved upon long-term follow-up, although some have suggested that certain histologic features may distinguish the two entities (114).
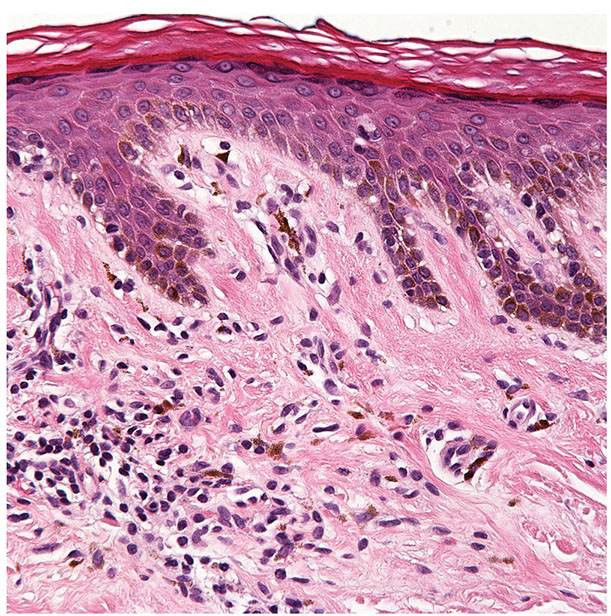
Figure 31-28 Hypopigmented mycosis fungoides. This variant is characterized by dermal pigment incontinence as well as the presence of an atypical T-cell infiltrate (which is subtle in this example).
Ichthyosiform mycosis fungoides. Another rare variant of MF, ichthyosiform MF presents with lesions that are pruritic and ichthyosiform, often with excoriations. There may be accompanying comedo-like areas and/or follicular keratotic papules. Microscopically, the ichthyosiform areas show typical features of MF with compact orthohyperkeratosis and hypergranulosis, whereas the follicular papules exhibit cystic dilatation of hair follicles with keratotic plugs and folliculotropic atypical lymphocytes (115).
Mycosis fungoides palmaris et plantaris. This variant defines cases where the MF lesions are limited to, predominantly affect, or initially present on the palms and/or soles (116). The lesions may present as annular hyperpigmented patches and plaques (117), vesicles, pustules (118,119), hyperkeratotic/verrucous plaques (120,121), psoriasiform plaques, ulceration (122), and nail dystrophy. Regardless of the associated variable secondary epidermal changes that may complicate the clinical differential and microscopic interpretation, the presence of histopathologic findings of classical MF and clonal rearrangement of the TCR genes will help in reaching the correct diagnosis.
Poikiloderma vasculare atrophicans. This is a reaction pattern that may occur in lesions of MF, although it is not synonymous with T-cell lymphoma and may be seen in other settings. Early lesions may involve either large plaques or small papules arranged in a netlike configuration. Lesions show erythema, mild scaling, epidermal thinning, mottled hypo- and hyperpigmentation, and telangiectasia, and the clinical picture may resemble changes of radiodermatitis. Histopathology discloses epidermal atrophy; epidermal infiltration by atypical lymphocytes; a variable band of lymphocytes, histiocytes, and melanophages in the papillary dermis; and ectasia of superficial dermal vessels (Fig. 31-29). The finding of atypical lymphocytes, potentially supported by a characteristic immunophenotypic and molecular profile for MF, should assist in separating this rare variant from other causes of acquired poikiloderma, including dermatomyositis and lupus erythematosus.
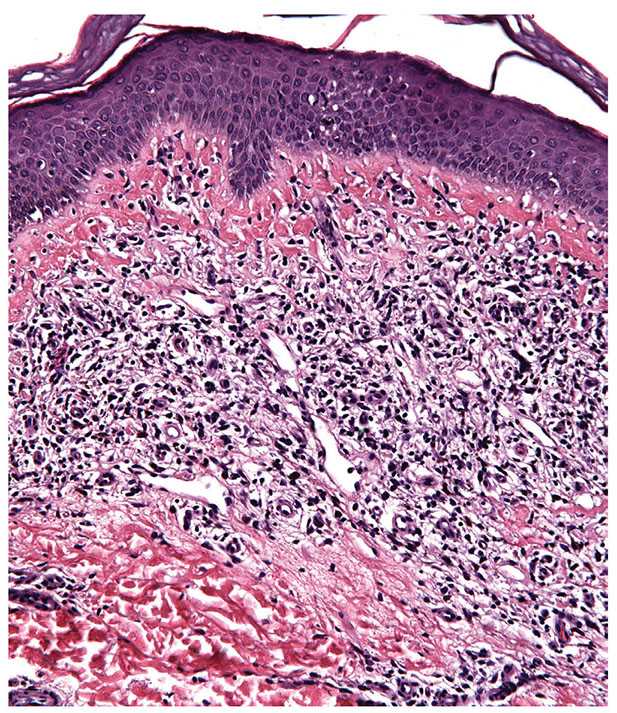
Figure 31-29 Poikiloderma vasculare atrophicans. This reaction pattern may occur in MF and is characterized by epidermal atrophy, epidermal infiltration by atypical lymphocytes, and a variable band of lymphocytes, histiocytes, and melanophages in the papillary dermis.
Pustular mycosis fungoides. Pustular MF presents as pustular eruptions that may be limited to the palmoplantar areas (MF palmaris et plantaris) or may be generalized. Intraepidermal collections of atypical lymphocytes with neutrophils and eosinophils constitute the characteristic histopathologic findings (123). Production of high levels of interleukin-8 by the lymphoma cells has been implicated in the formation of pustular lesions (124).
Verrucous (hyperkeratotic) mycosis fungoides. Typical lesions are hyperkeratotic/verrucous and occur on lower extremities, face, and trunk (125). There may or may not be associated classical patches and plaques of MF. Histologically, lesions are characterized by classic features of MF in a background of epidermal hyperplasia with hyperkeratotic scale (126,127).
Histogenesis. The putative cell of origin in MF is believed to be a memory T cell that expresses the skin-homing molecule CLA. Studies have failed to define a single genetic pathway of lymphogenesis, suggesting that the disease is genetically heterogeneous. For example, a large meta-analysis of mRNA gene expression data in tumor-stage MF demonstrated that 718 genes were statistically significantly more highly expressed in MF compared to normal or inflamed skin (128). A number of pathways have been implicated in disease pathogenesis including those involved in the regulation of apoptosis, cell-cycle progression, and gene transcription.
Differential Diagnosis. The practical differential diagnosis of classical epidermotropic MF involves primarily reactive T-cell infiltrates with exocytosis or an abnormal papillary dermal homing pattern (Table 31-5). True dermatitis may show lymphoid exocytosis and development of Langerhans cell microgranulomas that may superficially resemble Pautrier microabscesses. However, the microgranulomas are generally associated with spongiosis or evidence of cytotoxic epidermal injury, and CD1a positivity by immunohistochemistry (129). Adnexotropic lesions must be separated from forms of dermatitis primarily centered about hair follicles and sweat glands. The finding of uniformly atypical epitheliotropic lymphocytes and their tendency to be predominantly CD4+ by immunohistochemistry should assist in this differential. Careful clinical history is required to exclude the possibility of a drug-mediated skin eruption secondary to immune dysregulation that may be morphologically, immunophenotypically, and even genotypically difficult to differentiate from MF. Clearing of the skin lesions following discontinuation of the offending drugs or lack of correlation of TCR gene rearrangements among multifocal lesions is also helpful. Other common histologic mimickers of MF include persistent arthropod bite reaction, secondary syphilis, nodular scabies, actinic reticuloid, fungal infection, lichen sclerosus in the early inflammatory stage, lichen striatus, lichenoid keratosis, pigmented purpuric dermatitis, connective tissue disorders, inflamed vitiligo, and regressed melanoma (130).
It is possible that very early evolutionary stages of MF involve a dysplastic phase in tumor progression. Such lesions may fulfill some but not all of the features required for a diagnosis of patch-stage disease. The authors generally refer to such lesions as being compatible with possible evolving T-cell dyscrasia, and recommend close follow-up and interval biopsy sampling to assess disease evolution.
Late-stage disease, where epithelial involvement is minimal to absent and cytologic features may be higher grade, must be distinguished from B-cell lymphomas involving the skin, particularly T-cell-rich types.
A well-described diagnostic dilemma is the distinction between MF and primary cutaneous CD30+ lymphoproliferative disorders, including lymphomatoid papulosis (LyP) or primary cutaneous anaplastic large-cell lymphoma (cALCL). On pure histologic grounds, type B LyP mimics the bandlike infiltrate and epidermotropism of small- to medium-sized cells seen in MF, and type D LyP demonstrates striking epidermotropism that is morphologically similar to localized pagetoid reticulosis. Large, anaplastic CD30+ cells may be seen in both MF with LCT and cALCL. It cannot be overemphasized that incorporation of clinical history is essential for diagnosis. Recently, 6p25.3 IRF4/DUSP22 rearrangements have been described in approximately a quarter of cALCL cases and rarely in transformed MF (131,132).
Principles of Management. MF is generally considered to be incurable, although many patients will show an indolent course. Treatment options are tailored to the stage of disease. Therapy for early-stage lesions includes topical agents (corticosteroids, chemotherapy, bexarotene), phototherapy (psoralen plus UV-A or UV-B radiation), systemic bexarotene, and/or radiotherapy. More advanced disease (high-stage or transformed), or nonresponsive early-stage disease will require additional treatments such as systemic chemotherapy, extracorporeal photopheresis, interferon-α, and/or immunologic agents. Allogeneic stem cell transplant may also be considered for aggressive disease (133).
Sézary Syndrome
SS is a rare, aggressive disease classically characterized by a triad of generalized redness and scaling of the skin (erythroderma), lymphadenopathy, and the presence of clonally related neoplastic T cells in the skin, lymph nodes, and peripheral blood. However, the recent international consensus statement for MF and SS provides updated criteria for SS excluding the requirement of lymphadenopathy, specifically (a) confluence of erythema covering greater than or equal to 80% body surface, (b) a T-cell clone within the peripheral blood, and (c) a high blood tumor burden, meaning greater than or equal to 1,000/μL Sézary cells, a CD4:CD8 ratio greater than or equal to 10:1, or atypical CD4+ cells in the blood (CD4+CD7− cells ≥40% or CD4+CD26− cells ≥30%) (73,134). MF and SS are distinct entities with different postulated cells of origin (135) and underlying genetics (136). The term Sézary syndrome is restricted to cases without preceding MF lesions.
Clinical Summary. Patients generally present with diffuse skin erythema and lymphadenopathy. There may also be itching, hair loss, nail dystrophy, eye changes, and hyperkeratosis affecting the skin of the palms and soles. In addition to lymph node, skin, and blood involvement, malignant T cells may also infiltrate viscera in late stages, although bone marrow is often spared (137). The prognosis is poor, with a median survival between 2 and 4 years (2). Of note, patients with an established diagnosis of MF who develop erythroderma are considered to have erythrodermic MF, not SS, although these cases can be grouped under the name of erythrodermic CTCL (138).
Histopathology. Affected skin may show changes similar to those of patch/plaque-stage MF, although the infiltrate is often sparse and epidermotropism is less prominent (Fig. 31-30A). Similar to MF, the neoplastic T cells are generally CD4+ T cells that frequently show loss of CD7. Other aberrant T-cell phenotypes are common, often with loss of T-cell markers such as CD2, CD3, and CD5; rare CD8+ variants have been described. One study suggested that the follicular helper T-cell marker programmed death-1 (PD-1) is differentially expressed in SS, with staining seen in greater than 50% of cells in 24 of 27 SS cases versus 8 of 60 MF cases (139). Involved lymph nodes are characterized by effacement of their architecture by infiltrates of atypical cells.
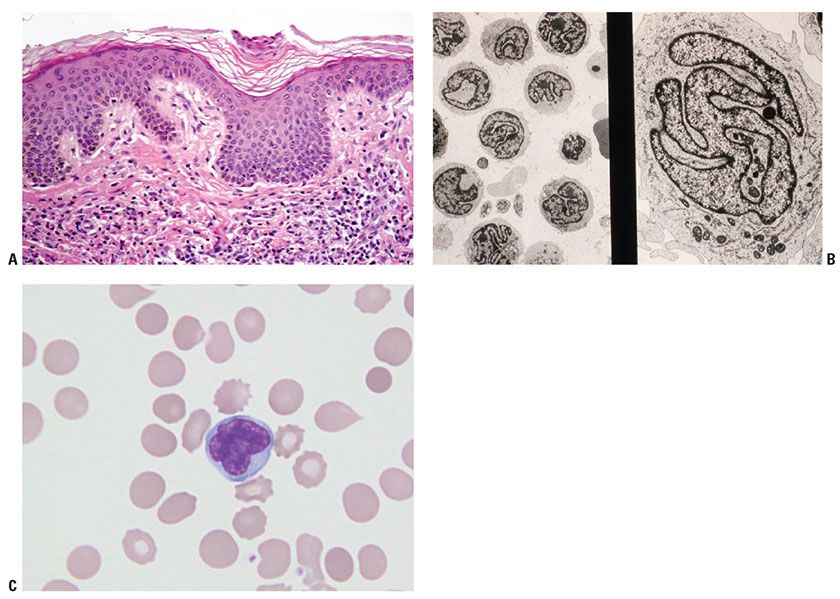
Figure 31-30 Sézary syndrome. A: There is a bandlike papillary dermal lymphoid infiltrate with minimal epidermotropism. B: Ultrastructural examination of peripheral blood buffy coat revealing numerous Sézary cells. C: High-magnification view of peripheral blood reveals the presence of an atypical lymphocyte with lobulated (“cerebriform”) nuclear contours (Wright stain).
The peripheral blood contains atypical lymphocytes with cerebriform nuclear contours, historically called Lutzner cells and larger Sézary cells (Fig. 31-30B, C). However, neither these morphologic features nor evidence of a clonal T-cell population in the blood are sufficient evidence of SS, as atypical, cerebriform lymphocytes are also found in the peripheral blood of otherwise healthy individuals and those suffering from inflammatory skin conditions, and clonal T-cell populations can be detected in the peripheral blood of otherwise healthy elderly individuals or those with autoimmune disorders (138,140–142). Therefore, either the presence of an immunophenotypically aberrant T-cell population in the blood (as described above) or demonstration of an identical T-cell clone in both the blood and the skin is required for the diagnosis.
Histogenesis. The putative cell of origin is thought to be the central memory T cell, which expresses markers that promote lymph node homing and peripheral blood circulation (135), although the underlying etiology is unknown.
Differential Diagnosis. SS must be differentiated from other dermatitic causes of erythroderma (e.g., atopic dermatitis, seborrheic dermatitis, pityriasis rubra pilaris, psoriasis, and drug reactions). This can be challenging on solely a morphologic basis because the histopathology of skin involvement in SS may be subtle, and additional features such as peripheral blood involvement will point toward a diagnosis of SS. Clinicopathologic correlation is needed to differentiate erythrodermic MF from SS. Patients with adult T-cell leukemia/lymphoma (ATLL) also have circulating atypical T cells and may present with skin lesions. However, the circulating lymphocytes tend to be polylobated (flowerlike) rather than cerebriform, and the cutaneous lesions manifest as multiple nodules rather than diffuse erythroderma.
Principles of Management. Treatment for erythrodermic CTCL is similar to advanced-stage MF, although extracorporeal photopheresis demonstrates superior efficacy in erythrodermic CTCL compared to MF (143–145).
Primary Cutaneous CD30-Positive T-Cell Lymphoproliferative Disorders
The entity of primary cutaneous CD30+ T-cell lymphoproliferative disorders encompasses a spectrum of diseases characterized by indolent clinical behavior and expression of CD30 including LyP and cALCL (146). Lesions that cannot be placed into one of these categories at initial diagnosis are considered borderline (1). Definitive diagnosis can be challenging on histologic grounds and requires careful clinical correlation.
Lymphomatoid Papulosis
Clinical Summary. LyP typically develops as generalized juicy red papules and nodules on the trunk and extremities that regress within weeks to several months (Fig. 31-31), resulting in mild scarring (147). The papules or nodules are each usually less than 1 cm in diameter, and ulceration may occur. Lesions are often at different stages of development resulting in a heterogeneous clinical appearance. The disease is typically chronic and can affect patients for months to years. LyP occurs most frequently in middle-aged adults (M:F 2–3:1), although it may also be seen in children. Five to 10% of cases may involve regional (draining) lymph nodes (148). A concurrent lymphoproliferative disorder such as CHL, cALCL, systemic ALCL, or MF may be present in up to 20% of cases (148–152). Although clonality can often be demonstrated in lesions of LyP, it is not clinically considered to be a malignant disorder.
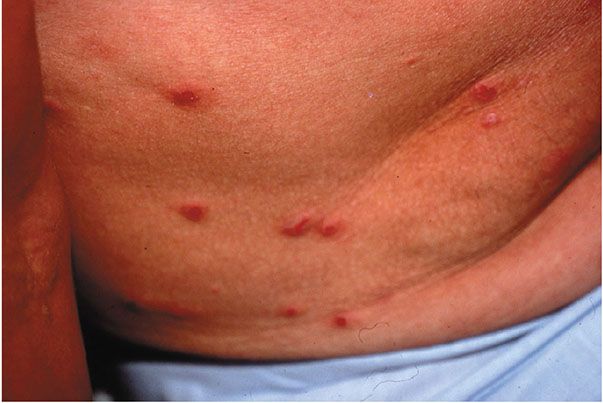
Figure 31-31 Lymphomatoid papulosis. Lesions clinically present as multiple, juicy red papules and nodules.
Histopathology. LyP is characterized by a cutaneous infiltrate of large, atypical CD30+ cells, which typically have an immunoblastic or anaplastic cytology. Multiple patterns have been described, and may occur within the same patient. LyP type A is the prototypical pattern typified by a wedge-shaped infiltrate extending from the superficial into the mid- and deep dermis (Fig. 31-32A). The infiltrate is composed of marked inflammatory component (small lymphocytes, histiocytes, neutrophils, and eosinophils) as well as an admixed minority population of scattered or clustered atypical large lymphocytes, which can be Reed–Sternberg–like (Fig. 31-32B–D). Type B mimics MF with small- to medium-sized cerebriform cells forming a bandlike superficial dermal infiltrate and exhibiting epidermotropism. Type C lesions show a monotonous population of large cells without a significant inflammatory component similar to cALCL, but clinically behave like LyP, hence their classification as such. Type D LyP recapitulates the striking epidermotropism of pagetoid reticulosis and the cytotoxic immunophenotype of primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma (CD8+, TCR βF1+, and TIA-1+ and/or granzyme B+) (153–155), although the presence of strong, uniform CD30 expression and the clinical picture support the diagnosis of LyP (6). Most recently, the term LyP type E has been proposed to describe an angioinvasive variant characterized by ulcerative/necrotic lesions due to an underlying angiocentric and angiodestructive infiltrate of small- to medium-sized atypical lymphocytes frequently expressing CD8 (156,157). In general, the atypical CD30+ cells show expression of CD4 with variable loss of CD2, CD3, or CD5. CD8+ cases are rare, typically seen in pediatric cases or types D and E. Expression of cytotoxic molecules (granzyme B, TIA-1) may be seen in the CD30+ cells (158). The small- to medium-sized atypical cells in LyP type B may or may not express CD30.
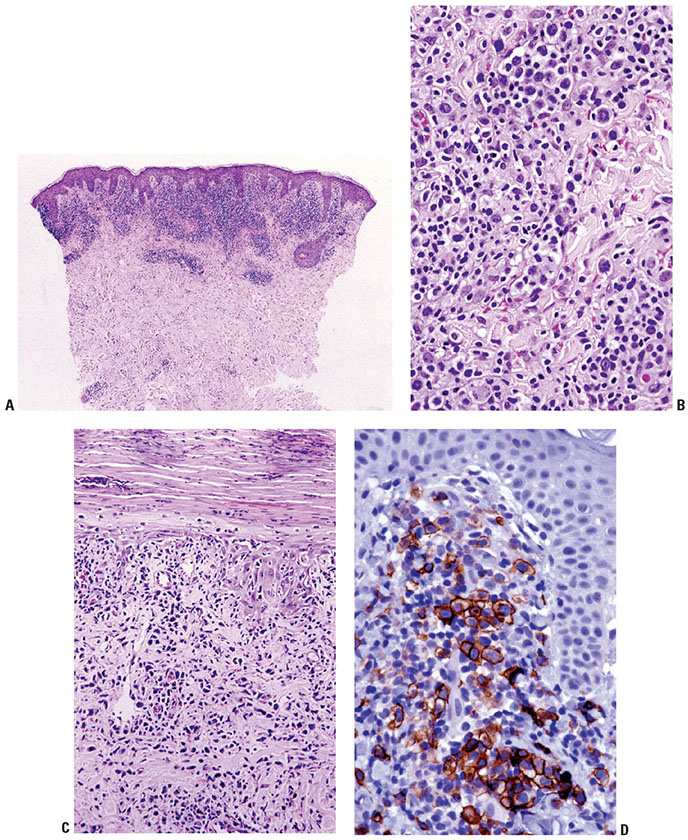
Figure 31-32 Lymphomatoid papulosis. A: Low power of localized infiltrate in superficial and mid-dermis, forming what will become a wedge-shaped angiocentric architecture. B: Variably atypical lymphoid cells in the dermis. C: Epidermal infiltration with associated scale abnormalities and neutrophil infiltration. D: CD30+ cells are numerous within the superficial dermal component of the infiltrate.
Histogenesis. The cell of origin is thought to be a skin-homing T cell. Until recently, recurrent genetic abnormalities have not been described in LyP, and to date no chromosomal abnormalities defining the histopathologic type A through E patterns have been identified. However, a small series of spontaneously resolving, localized cutaneous lesions in elderly patients characterized by rearrangements of the IRF4-DUSP22 locus on chromosome 6p25.3 has been reported (159). These cases were felt to best fit in the category of LyP given the strong expression of CD30 and self-resolving course, although they did not fit perfectly into one of the five described patterns. Previously, rearrangements at 6p25.3 have been found to be a specific but insensitive indicator of cALCL, present in approximately 25% of cases (6,131,132,160). Thus, the presence of this rearrangement in both LyP and cALCL provides further evidence that entities within the category of indolent primary cutaneous CD30+ T-cell lymphoproliferative disorders represent a related spectrum of disease. In terms of the spontaneous resolution characteristic of LyP, studies suggest that this may be secondary to enhanced apoptosis (161–164). TCR gene rearrangements are detected in approximately 60% of cases of LyP (152).
Differential Diagnosis. In general, clinical correlation will be essential in distinguishing the various patterns of LyP from morphologically similar entities as mentioned above, as well as from transformed, CD30+ MF. Interestingly, rare cases have been reported in which MF and primary cutaneous CD30+ lymphoproliferative disorders coexist (148,150,165), which may predict an improved prognosis compared to MF alone (74). Another entity to consider is pityriasis lichenoides et varioliformis acuta (PLEVA), which presents with papulonecrotic lesions; distinction may be particularly difficult in cases with numerous CD30+ cells (166). Other reactive, self-resolving conditions, including arthropod bites, drug eruptions, and viral infection (e.g., herpes simplex virus 1 and 2), should also be considered (167–169).
Principles of Management. LyP may not require any therapy, particularly in cases where the extent of disease is limited. Patients with disseminated or disruptive lesions may utilize phototherapy or low-dose methotrexate (146). Systemic chemotherapy will only provide short-term effectiveness and should not be used in this clinical setting. Long-term clinical follow-up is advised given the relationship of LyP with other lymphoproliferative disorders such as MF.
Primary Cutaneous Anaplastic Large-Cell Lymphoma
Clinical Summary. Skin lesions of cALCL consist of one or more localized, rapidly growing, frequently ulcerated reddish-brown nodules or occasionally papules (72,148). Partial regression of untreated lesions may occur, but complete spontaneous resolution as seen in LyP is unusual. Multifocal disease is less common, and may be associated with relapse following therapy. Nonetheless, cALCL is associated with an excellent prognosis with an estimated 5-year survival of greater than 90% (170–172). There may be involvement of regional, draining lymph nodes, but this does not affect prognosis (148). Extracutaneous dissemination occurs in approximately 10% of patients and may be more common in patients who present with multifocal disease (173).
Histopathology. cALCL is characterized by nodular or diffuse infiltrates of large, anaplastic cells that involve the dermis and occasionally the subcutis (Fig. 31-33A, B). Morphologically, biopsies of cALCL may closely resemble those of systemic ALCL with secondary skin involvement, although Reed–Sternberg–like cells and highly pleomorphic, multinucleated giant cells may be more numerous in the latter. The large, atypical cells should account for the majority of the infiltrate, although a background of secondary inflammatory elements may be observed and occasionally eosinophilia is marked. Necrosis and/or angioinvasion may be seen. Marked epidermal hyperplasia, sometimes with keratoacanthoma-like features; epidermal invasion by atypical cells; and ulceration are often present. Greater than 75% of the infiltrate is, by definition, positive for CD30 (Fig. 31-33C). The cells are typically CD4+, and frequently show expression of cytotoxic granule proteins (granzyme B, TIA-1) (158,174). Aberrant T-cell phenotypes with loss of CD2, CD3, CD5, and/or CD7 may be observed, and CD8 expression rather than CD4 expression may be seen. EMA and ALK1 are negative.
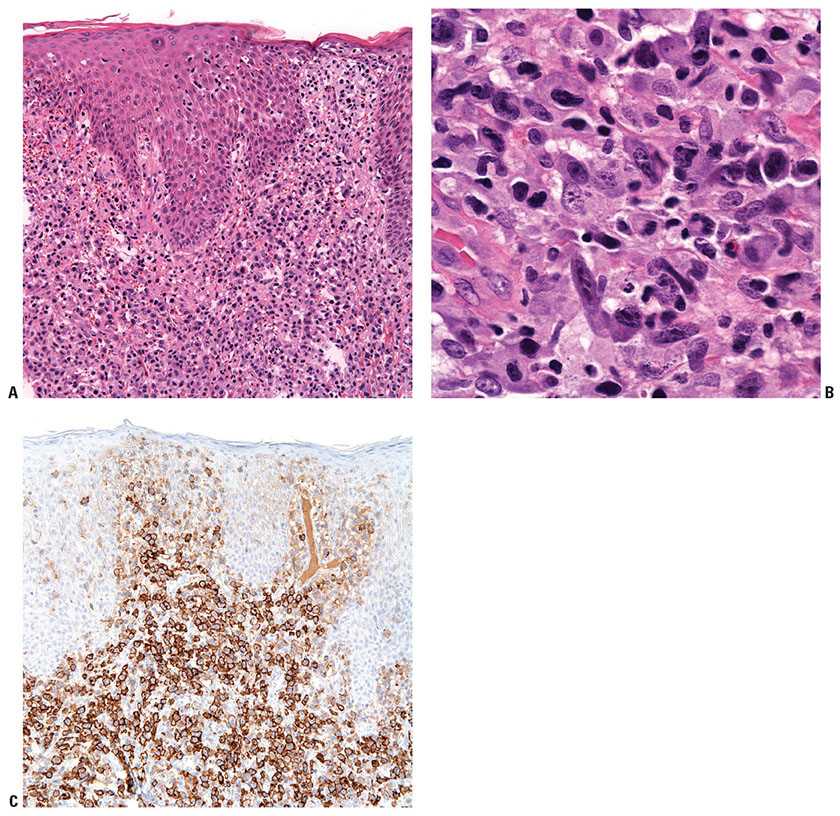
Figure 31-33 Primary cutaneous anaplastic large-cell lymphoma. A: Tumoral infiltrate of large, atypical lymphocytes with associated incipient epidermal erosion. B: High magnification of the large, anaplastic lymphocytes. C: Strong CD30 expression in the atypical lymphocytes.
Histogenesis. All forms of primary cutaneous CD30+ T-cell lymphoproliferative disorders are believed to be derived from an activated skin-homing T cell. Most cases of cALCL show clonal rearrangements of the TCR genes. In contrast to some cases of systemic ALCL, translocations involving the ALK (anaplastic lymphoma kinase) gene are not detected (175). Rearrangements at 6p25.3 have been found to be a specific but insensitive indicator of cALCL, present in approximately 25% of cases (6,131,132,160). Rearrangements at this locus have also been described in peripheral T-cell lymphomas (160), and more recently in a subset of LyP-like cases (159).
Differential Diagnosis. In general, demonstration of ALK1 expression in a cutaneous CD30+ T-cell lymphoma with anaplastic morphology should suggest a diagnosis of secondary involvement by systemic ALCL, ALK+. However, rare cases of CD30+ cutaneous tumors with ALK1 immunohistochemical expression and/or ALK translocations arising in patients without evidence of systemic disease (even with long-term follow-up) have been described (6,176–178). Nonetheless, these cases are the exception and not the rule, and careful staging should always be performed in patients with CD30+, ALK1+ cutaneous tumors. Likewise, careful clinical correlation including staging is needed to distinguish cALCL from other CD30+ tumors with considerably worse prognoses including secondary cutaneous involvement by ALK1− systemic ALCL, LCT of MF, and other high-grade large-cell lymphomas that may also express CD30. Regional draining lymph nodes may be involved by cALCL, which may be morphologically indistinguishable from focal nodal involvement by CHL (179). Therefore, detection of CD30+ cells with anaplastic morphology in an isolated lymph node should prompt careful examination of the overlying skin to evaluate for the possibility of cALCL.
Principles of Management. Surgical excision or radiotherapy may be utilized for localized lesions, whereas systemic chemotherapy may be needed in multifocal disease.
Subcutaneous Panniculitis-Like T-Cell Lymphoma
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare lymphoma that primarily infiltrates subcutaneous tissue, demonstrates an αβ T-cell phenotype, and infrequently is associated with a hemophagocytic syndrome (180).
Clinical Summary. SPTCL tends to show an equal gender distribution and occurs mainly in adults, although childhood cases have been reported (181). Affected individuals generally present with symptoms related to formation of solitary or generalized deeply seated, variably sized erythematous cutaneous nodules often involving the extremities and/or trunk. Ulceration is uncommon, and extracutaneous spread is rare. Approximately 60% of patients present with systemic symptoms such as fever, fatigue, and weight loss, and a smaller number (approximately 20%) demonstrate evidence of a hemophagocytic syndrome. The disease has a protracted but indolent course with an overall 5-year survival of 82%. Notably, the overall survival of patients without a hemophagocytic syndrome is 91% compared to only 46% when a hemophagocytic syndrome is present (180).
Histopathology. There is a diffuse, variably cellular infiltrate involving both septa and lobules within the subcutis (Fig. 31-34A), with relative sparing of the overlying dermis and epidermis. Given this distribution pattern, a large biopsy or excision is needed to evaluate for involvement when this disease is suspected. The infiltrate is composed of small- to medium-sized lymphocytes with visible cytoplasm admixed with fewer large transformed cells with hyperchromatic nuclei. Often the initial biopsy, in which the more atypical elements are not well-represented, may appear deceptively banal. Cells characteristically show peripheral alignment (rimming) around individual adipocytes, although this finding is nonspecific (Fig. 31-34B). Fat necrosis, karyorrhexis, and associated infiltration of reactive histiocytes may also be noted. Necrosis, nuclear fragmentation, and vascular injury may all be observed. Although nonneoplastic small lymphocytes may be present, admixed plasma cells and eosinophils are not commonly observed. The malignant cells express a mature CD3+, CD4−, and CD8+ T-cell phenotype. CD30 and CD56 staining are consistently negative. EBER in situ hybridization is also always negative. The cells by definition demonstrate an αβ T-cell phenotype, and use of antibodies to TCR βF1 and to the TCR gamma chain constant region should be used in any suspected case of SPTCL to document the phenotype.
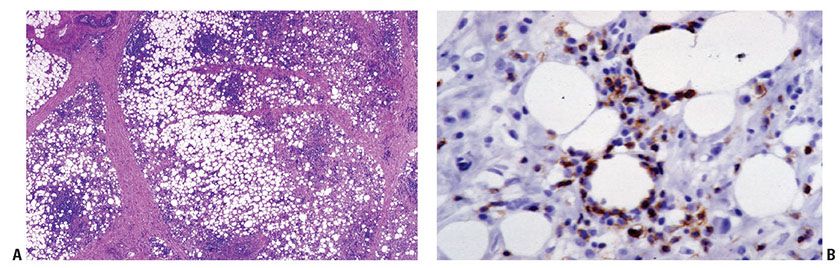
Figure 31-34 Subcutaneous panniculitis-like T-cell lymphoma. A: Lobular and septal pattern of subcutaneous infiltration. B: TIA-1 immunohistochemistry showing characteristic rimming around individual adipocytes.
Histogenesis. In keeping with their presumed derivation from a cytotoxic T cell, the neoplastic T cells also are reactive for granzyme, perforin, and TIA-1. Clonality can be demonstrated by TCR gene rearrangements. The mechanism underlying the association of hemophagocytic syndrome with SPTCL has not yet been determined.
Differential Diagnosis. SPTCL should be differentiated from benign panniculitis, such as lupus erythematosus panniculitis. The presence of plasma cells and lymphoid follicles with germinal centers and the lack of clonality in TCR gene rearrangement studies favor the latter. Extranodal NK/T-cell lymphoma, nasal type (ENKTCL), which frequently exhibits dermal in addition to subcutaneous involvement, is CD56+, demonstrates reactivity for EBER in situ hybridization and generally does not show TCR gene rearrangements. Demonstration of a γδ T-cell phenotype excludes SPTCL and most likely indicates a diagnosis of primary cutaneous γδ T-cell lymphoma, which may or may not express CD8, expresses for CD56, and has a poor prognosis (180,182).
Principles of Management. A definitive consensus on therapy for SPTCL is yet to be determined. Depending on the severity of disease (including the presence of a hemophagocytic syndrome), possible treatments include use of systemic steroids or other immunosuppressive therapies such as cyclosporine; radiation therapy for solitary lesions; or multiagent chemotherapy with or without autologous or allogeneic stem cell transplant.
Primary Cutaneous Gamma-Delta T-Cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGD-TCL) was previously characterized as SPTCL with a γδ T-cell phenotype (183). The WHO/EORTC classification defines this entity as a cutaneous lymphoma originating from a clonal proliferation of mature, activated γδ T cells with a cytotoxic phenotype and a generally aggressive clinical behavior.
Clinical Summary. PCGD-TCL affects adults without gender predilection. Typical lesions include disseminated indurated plaques and ulcerated/necrotic nodules/tumors, most commonly on the extremities and trunk; scaly patches are infrequent (184). Systemic involvement of mucosal and other extranodal sites is common, but the lymph nodes, spleen, or bone marrow are generally spared (184,185). A hemophagocytic syndrome occurs in 50% of patients (180). The prognosis is disappointing even with systemic chemotherapy, with a 5-year overall survival of 11%, regardless of the presence or absence of a hemophagocytic syndrome (180). However, a distinct subset of patients has shown a more indolent course (186). It has been reported that patients with subcutaneous disease have a worse prognosis compared with those with epidermal or dermal infiltrates only (187), although no statistically significant difference in 2-year survival was seen in panniculitic or nonpanniculitic cases in a subsequent study (184).
Histopathology. The infiltrate may be epidermotropic, diffusely dermal, panniculitic, or show combinations of the above patterns either in one biopsy or in different specimens (Fig. 31-35A, B) (187,188). The cytomorphology is monomorphous with variably sized cells displaying irregular to pleomorphic nuclei and coarsely clumped chromatin (Fig. 31-35C). Angioinvasion is common, although the presence of large areas of necrosis secondary to angiodestruction is not a typical feature (189). Subcutaneous involvement may show rimming of adipocytes, similar to SPTCL. In cases associated with hemophagocytic syndrome, phagocytosis of blood cells by histiocytes is observed (185). The neoplastic cells commonly demonstrate a CD3+, CD2+, CD4−, CD5−, CD7+/−, CD8−, and CD56+ phenotype, although cases with CD8 expression are also seen (184). EBER in situ hybridization reactivity is generally not present, although rare cases have been described (184,190,191). By definition, the cells show a γδ T-cell phenotype, which can be demonstrated by reactivity with an antibody to TCR gamma chain constant region, which is now available for use on paraffin sections (previous antibodies could only be used for frozen section immunohistochemistry) (Fig. 31-36A, B).
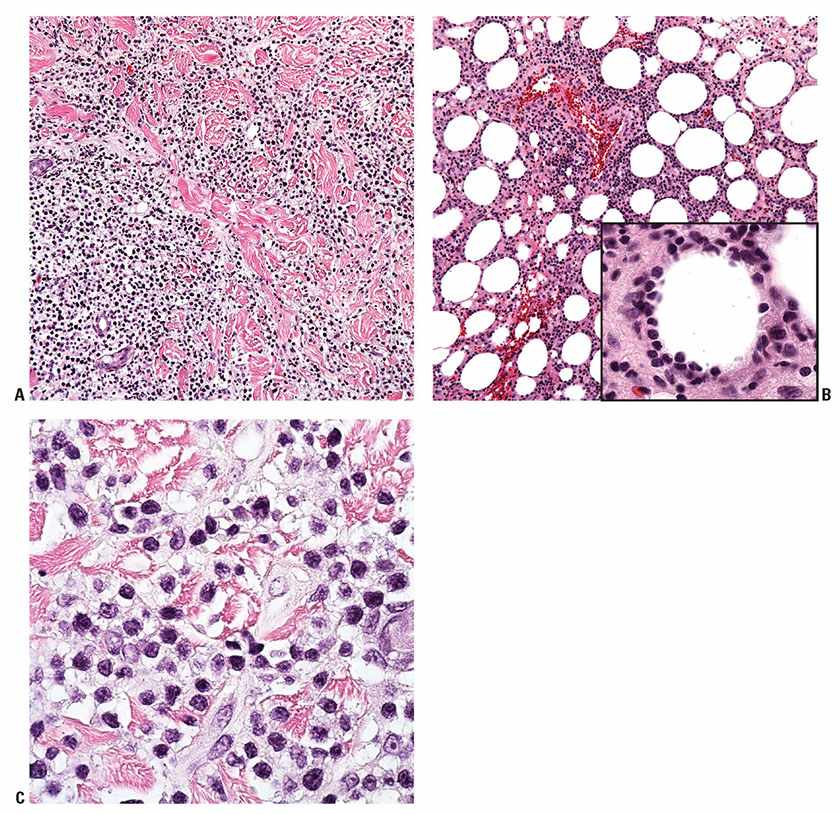
Figure 31-35 Primary cutaneous γδ T-cell lymphoma (PCGD-TCL). A: A diffuse infiltrate of neoplastic T cells invades the dermis in one of the patterns of PCGD-TCL. B: PCGD-TCL may also manifest as a panniculitic pattern with adipocyte rimming (inset) as is also seen in SPTCL. C: The cells may be large and pleomorphic but typically show condensed chromatin; blastic-appearing chromatin is not a common feature of this entity.
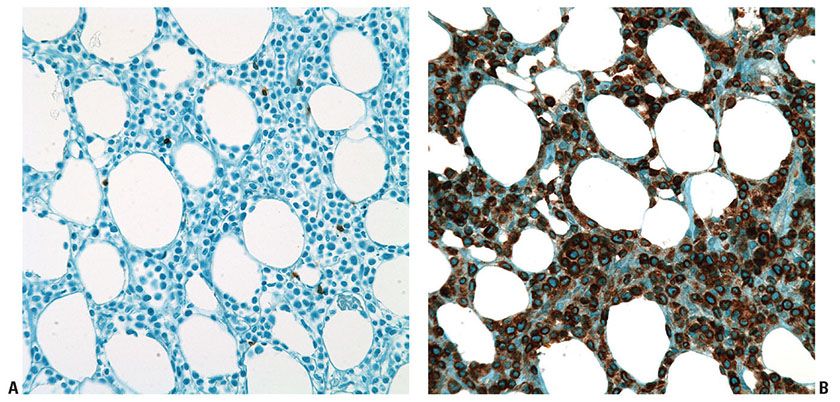
Figure 31-36 Primary cutaneous γδ T-cell lymphoma. A: Immunohistochemistry on paraffin sections using the TCR βF1 antibody demonstrates absence of reactivity in the infiltrate and highlights few admixed reactive T cells. B: Immunohistochemistry on paraffin sections using a monoclonal antibody to the TCR gamma chain constant region reveals strong reactivity within the infiltrate.
Histogenesis. Given the postulated origin from a mature and activated cytotoxic γδ T cell, there is strong expression of cytotoxic proteins (TIA-1, granzyme B, and perforin). Clonality can be demonstrated by TCR gene rearrangement studies.
Differential Diagnosis. Rare cases of MF or LyP type D with a γδ T-cell phenotype have been described, and clinical correlation is needed for distinction (192). Differentiation between ENKTCL and PCGD-TCL can pose a particular problem given that both diseases share clinical, morphologic, and immunophenotypic features. In most cases, distinction is straightforward now that an antibody is available to demonstrate the γδ T-cell phenotype. Additionally, TCR gene rearrangements favor PCGD-TCL, while diffuse positivity for EBER in situ hybridization as well as large areas of necrosis associated with angiodestruction favors ENKTCL. However, cases showing overlapping features have been described (193), and indeed the two entities demonstrate similar genetic signatures (194). Therefore, in rare cases, definitive categorization may not be possible. Both SPTCL and primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma demonstrate an αβ T-cell phenotype and are generally CD56−.
Principles of Management. Systemic chemotherapy is typically utilized, although the disease is frequently resistant to treatment.
Primary Cutaneous CD8-Positive Aggressive Epidermotropic Cytotoxic T-cell Lymphoma (Provisional Entity)
This provisional entity (herein referred to as PCCD8AC-TCL) is characterized by a proliferation of epidermotropic CD8+ cytotoxic T cells and an aggressive clinical course (195–197). Prior to the recognition of this category, patients with this tumor were classified as having generalized pagetoid reticulosis (Ketron–Goodman disease) or an aggressive MF.
Clinical Summary. Affected patients are typically middle-aged to elderly, with a median age of 53 years and a male-to-female ratio of 1.4:1.0 (195). Clinically, patients present with localized or disseminated eruptive papules, nodules, or tumors with central ulceration and necrosis or hyperkeratotic patches and plaques (195,198). Spread to other visceral sites, such as the lung, testis, central nervous system, and oral mucosa, has been documented, but nodal involvement is unusual (195). The clinical course is aggressive, with rapid progression and a median survival of less than 32 months (2,195)
Histopathology. PCCD8AC-TCL lesions are characterized by variably sized, pleomorphic T lymphocytes forming heterogeneous patterns including lichenoid, nodular, or diffuse infiltrates, commonly with marked epidermotropism (Fig. 31-37A, B). In advanced lesions, the epidermotropism may be less pronounced. The epidermis may be acanthotic or atrophic with variable numbers of apoptotic keratinocytes and spongiosis. Associated ulceration, necrosis, or blister formation may be observed. Adnexal invasion/destruction is frequently present, while angioinvasion is less common. The atypical lymphoid infiltrate may be accompanied by a variable number of reactive histiocytes and dendritic cells as well as rare eosinophils and plasma cells (195,199). The neoplastic cells display a CD3+, CD4−, CD8+, CD7−/+, CD45RA+, CD45RO−, granzyme B+, perforin+, and TIA-1+ phenotype, characteristic of cytotoxic T cells, and show an αβ T-cell phenotype. CD2 and CD5 expression are commonly lost (Fig. 31-38A–D). Generally CD56 and CD30 are negative, and there is no reactivity in tumor cells for EBER in situ hybridization. The Ki67 proliferation index is high.
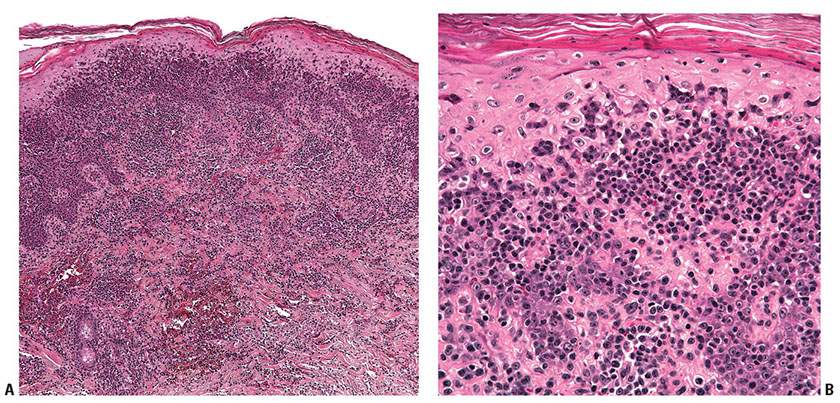
Figure 31-37 Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma. A: Scanning magnification reveals neoplastic cells forming a dermal interstitial pattern as well as a dense infiltrate in the overlying epidermis. B: Higher magnification reveals the small- to intermediate-sized atypical lymphocytes permeating the epidermis in a pagetoid pattern.
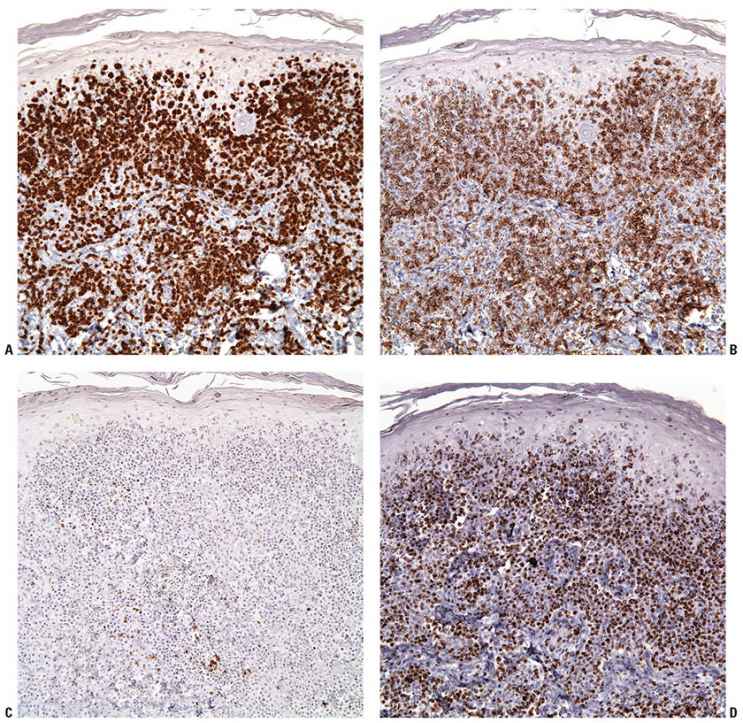
Figure 31-38 Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (PCCD8AC-TCL). A–D: Immunohistochemistry studies reveal the characteristic immunophenotype of PCCD8AC-TCL including expression of CD3 (A), CD8 (B), loss of CD2, (C) and expression of the cytotoxic molecule perforin (D).
Histogenesis. The postulated cell of origin is a skin-homing, CD8+, cytotoxic αβ T cell. TCR gene rearrangement studies are typically positive.
Differential Diagnosis. Differentiation from other types of epidermotropic CTCLs expressing a CD8+ cytotoxic T-cell phenotype, such as LyP type D and rare cases of MF (particularly tumor stage or transformed), is largely based on clinical presentation and disease course. In addition, LyP will show expression of CD30, which has only rarely been reported in PCCD8AC-TCL (6). Although the clinical presentation is indistinguishable from PCGD-TCL, tumors of PCCD8AC-TCL are of an αβ T-cell phenotype.
Of note, several recent reports describe indolent proliferations of small- to medium-sized, pleomorphic, nonactivated cytotoxic CD8+ T cells of the ear/face or at acral sites (200,201). While these lesions share morphologic and phenotypic overlap with PCCD8AC-TCL (CD8+; expression of TIA-1), they can be distinguished on a pathologic basis by the absence of striking epidermotropism (in fact, a Grenz zone is frequently observed), variable expression of perforin and granzyme B in accordance with the nonactivated state, and low Ki67 proliferation index.
Principles of Management. Given the rarity of this entity, treatment guidelines are not well-established.
Related posts:
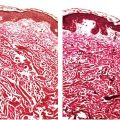

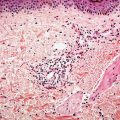
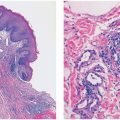
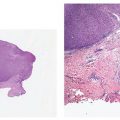
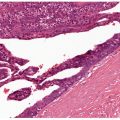
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


