Craniosynostosis Syndromes
Scott P. Bartlett
Christopher A. Derderian
Craniosynostosis, or premature closure of cranial vault and cranial base suture, can involve any suture. Those most commonly observed, in order of decreasing frequency, are sagittal, coronal, metopic, and lambdoidal (Chapter 22). In simple craniosynostosis, one suture is prematurely fused. In multiple-suture synostosis, two or more sutures are prematurely fused. Craniosynostosis can occur as an isolated event resulting in non-syndromic craniosynostosis, or it can occur in conjunction with other anomalies in well-defined patterns that make up clinically recognized syndromes. Syndromic craniosynostosis is most often genetic in nature, and patterns of autosomal dominant, autosomal recessive, and X-linked inheritance have been observed. More than 90 reported syndromes are associated with craniosynostosis, with most involving associated anomalies of the limbs, ears, and cardiovascular system.
The Apert, Crouzon, Pfeiffer, Saethre-Chotzen, and Muenke syndromes represent the more commonly identified craniosynostosis syndromes seen by plastic surgeons. These familial craniosynostosis syndromes share many common features, including midface hypoplasia, cranial base growth abnormalities, abnormal facies, and limb abnormalities. In fact, the craniofacial features are clinically similar among the various syndromes so that the anomalies of the hands may be the differentiating clinical feature between the various syndromes. Although it is clear that synostosis of the cranial sutures is significantly involved in the development of the abnormal craniofacial features in these syndromic children, there probably exists a mesenchymal defect in the cranial base that also contributes to the craniofacial deformity.
The exact etiology of the craniosynostosis in these syndromic children remains unclear. Advances in molecular genetics provide insights into a possible link between mutations identified in fibroblast growth factor receptor (FGFR) genes and several autosomal dominant skeletal disorders. Fibroblast growth factors participate in the regulation of cell proliferation, differentiation, and migration and play a role in controlling normal bone morphogenesis via complex cell-signaling pathways. The transduction of a fibroblast growth factor signal to the cytoplasm is mediated by a group of transmembrane tyrosine kinase receptors known as the FGFRs. Mutations in three of the four known FGFR genes located on chromosomes 8, 10q, and 4p have been identified in the Pfeiffer, Apert, Crouzon, Muenke, and Jackson-Weiss syndromes. Achondroplasia, a skeletal disorder that causes the most common form of short-limb dwarfism, is also linked to a mutation in the FGFR complex. The Pfeiffer syndrome is linked to a mutation in both the FGFR1 and FGFR2 genes, whereas the Crouzon and the Apert syndromes are linked to mutations in the FGFR2 alone. The complexity of this system is highlighted by the observation that two distinct phenotypes of Apert syndrome are caused by missense substitutions in the base sequence for adjacent amino acids in the FGFR2 gene resulting in a higher incidence of cleft palate in one (S252W) and more severe syndactylism in the other (P253R).
CROUZON SYNDROME (ACROCEPHALOSYNDACTYLY TYPE II)
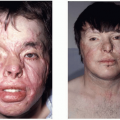
Crouzon syndrome is characterized by premature fusion of calvarial sutures, midface hypoplasia, shallow orbits, ocular proptosis, and FGFR-2 receptor mutation (Figure 23.1). The clinical features were first described by Crouzon, a French neurologist, in 1912. The pattern of inheritance is autosomal dominant. The reported frequency is 1 in 25,000 live births. The variability in expression of the dominant features that make up Crouzon syndrome is widely recognized, and mild deformities in a previously undiagnosed parent of a more severely affected child is a common scenario.
Premature fusion of both coronal sutures, resulting in a brachycephalic head, is the most common calvarial deformity, but scaphocephaly and trigonocephaly, as well as the cloverleaf skull deformity, have been observed, as has “normocephalic” pancraniosynostosis. The craniosynostosis is typically complete by 2 to 3 years of age, but often the sutures are fused at birth. The cranial base sutures are frequently involved, resulting in maxillary and midface hypoplasia. The maxillary hypoplasia is evidenced by a reduced dental arch width and a constricted, high palatal arch. Normal or near-normal mandibular growth leads to a class III malocclusion. The midface hypoplasia is reflected in the shallow orbits with exorbitism, which is a consistent finding and can result in exposure conjunctivitis or keratitis. Exorbitism can be so severe that herniation of the globe through the eyelids may occur, requiring immediate reduction. Acuity problems, strabismus, and hypertelorism have all been reported. A conductive hearing deficit is not uncommon. The defining characteristic of Crouzon syndrome is that no commonly reported limb anomalies are present in this population of syndromic craniosynostosis patients.
Several reports have identified Crouzon syndrome as carrying a higher risk of elevated intracranial pressure (ICP) than other forms of syndromic craniosynostosis, with one study demonstrating a 65% incidence of elevated ICP and the remainder borderline elevated. A retrospective review of suture patency in patients with syndromic craniosynostosis may offer some insight. The review demonstrated that patients with Crouzon syndrome demonstrated earlier closure of the lambdoid and sagittal sutures (median 6 and 21 months, respectively) and a 72% incidence of type I Chiari malformation as compared with patients with Apert syndrome with later suture closure (51 and 60 months, respectively) and a 2% incidence of type I Chiari malformation. We agree with others that these clinically significant characteristics are likely related and should be considered when planning the sequence for surgical treatment of these patients.
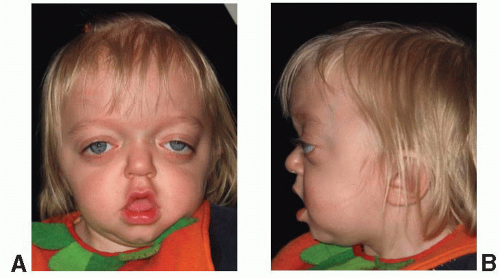 FIGURE 23.1. Crouzon syndrome in a young female. Note the midface hypoplasia, shallow orbits, and ocular proptosis. A. Frontal view. B. Profile view. |
APERT SYNDROME (ACROCEPHALOSYNDACTYLY TYPE I)
Apert, in 1906, described a syndrome characterized by craniosynostosis, exorbitism, midface hypoplasia, and symmetric syndactyly of both hands and feet. Recently mutations in genes coding for FGFR-2 have been identified in patients with Apert syndrome (Figure 23.2). The incidence is reported to be between 1 in 100,000 and 160,000 live births. Most cases are sporadic, although several cases with autosomal dominant transmission have been reported. The cranial vault deformity in these patients is variable, but most often presents as a short anteroposterior dimension with craniosynostosis involving the coronal sutures resulting in a turribrachycephalic skull, with a large anterior fontanelle. The typical craniofacial appearance includes a flat, elongated forehead with bitemporal widening and occipital flattening. The midface hypoplasia is accompanied by orbital proptosis, downslanting palpebral fissures, and hypertelorism. The nose is downturned at the tip, the bridge is depressed, and the septum is deviated (parrot beak deformity).
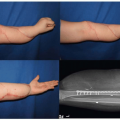
The maxillary hypoplasia results in a class III malocclusion with an anterior open bite, and commonly results in significant enough airway compromise to warrant tracheostomy. All patients have a high arched palate and 30% have an associate cleft palate. The hand syndactyly, which is pathognomic for the condition, most often involves fusion of the index, middle, and ring fingers, resulting in mid-digital hand mass; the thumb and little fingers may also be joined to the mid-digital mass (Figure 23.3). When the thumb is free, it is broad and deviates radially due to an accompanying delta phalanx. In the feet, the syndactyly also usually involves the second, third, and fourth toes. These hand anomalies are so severe and functionally debilitating that referral to a hand surgeon with special expertise in this area is essential. An extensive review of central nervous system problems in patients with Apert syndrome shows an increased incidence of delayed mental development, but many of these patients develop normal intelligence. Acne vulgaris is another characteristic feature seen during adolescence in over 70% of patients.
Perhaps our best understanding of the natural history of raised ICP in Apert syndrome comes from the Great Ormond Street Hospital data on the expectant management of their patients. Their protocol is to offer cranial vault expansion only in the setting of confirmed elevation of ICP. Raised ICP developed in 83% (20/24) of patients, 50% in the first year of life, with the average age of onset at 18 months (range 1 month to 4 years 5 months). Thirty-five percent of those who were treated successfully for their first episode of elevated ICP went on to develop a second episode on average 3 years 4 months later.
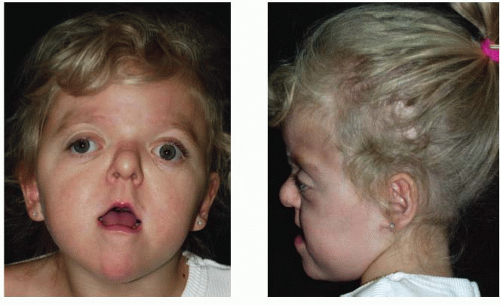 FIGURE 23.2. Apert syndrome in a 2-year-old female. Note the severe midface hypoplasia, elongated forehead with temporal widening, brachycephaly, and beaked nose. |
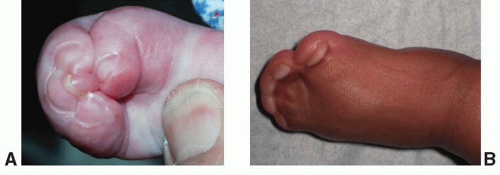 FIGURE 23.3. A and B demonstrate the characteristic hand and foot syndactyly, which are pathognomic for Apert syndrome. This complex syndactyly most often involves fusion of the second, third, and fourth fingers, resulting in mid-digital hand mass, but the first and fifth fingers may also be joined to the mid-digital mass. In the feet, the syndactyly also usually involves the second, third, and fourth toes. |
PFEIFFER SYNDROME (ACROCEPHALOSYNDACTYLY TYPE V)
This syndrome was described by Pfeiffer in 1964 and consists of craniosynostosis, broad thumbs, broad great toes, and, occasionally, a partial syndactyly involving the second and third digits. Pfeiffer syndrome is linked to mutations in both FGFR-1 and FGFR-2, which confer less and more severe craniofacial dysmorphism, respectively. Symptoms vary, ranging from very mild to severe. The mode of inheritance is autosomal dominant. The craniofacial features are similar to those of Apert or Crouzon syndrome. The skull is turribrachycephalic secondary to the coronal and occasional sagittal synostosis (Figure 23.4). Maxillary hypoplasia with resulting midface deficiency leads to shallow orbits and exorbitism. Hypertelorism and downslanting palpebral fissures are also common. The nose is often downturned with a low nasal bridge. Intelligence is reported to be normal in the more common form of Pfeiffer syndrome. Broad thumbs and great toes are the hallmark of the syndrome, but the findings are frequently subtle. The partial syndactyly of the hands usually involves the index and middle fingers. A partial syndactyly of toes 2, 3, and 4 has also been noted. Cohen proposed a classification system that clusters patients into three types based upon their clinical findings and severity. Type I represents the “classic Pfeiffer” syndrome (features described above) which is milder than type II with a kleeblattschädel (cloverleaf) skull, and type III Pfeiffer syndrome is the most severely affected.
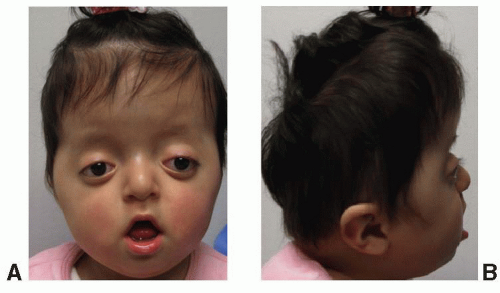 FIGURE 23.4. Pfeiffer syndrome in an infant female. A. Note the severe midface hypoplasia, exorbitism, mild hypertelorism, and turribrachycephalic skull. B. The profile view clearly demonstrates the abnormal brow-to-cornea relationships, concavity of the midface, and short nose. |
A recent review of 28 patients treated at a single institution reported that the Cohen subtypes distribution was type 1 61%, type II 25%, and type III 14%(Fearon). These patients underwent an average of 2.5 cranial vault procedures, 1.6 neurosurgical procedures, and 3.5 other operations. In addition to the challenging reconstructive needs of the patient with Pfeiffer syndrome, this study highlighted several functional considerations that should be aggressively treated or monitored in these patients including value in early placement of permanent tarsorrhaphies and supplementing these with temporary tarsorrhaphies at the time of cranial vault procedures, and high incidences of aural atresia (54%), conductive hearing loss (86%), need for tracheostomy (61%), hydrocephalus (68%), and Chiari malformations (82%).
SAETHRE-CHOTZEN SYNDROME (ACROCEPHALOSYNDACTYLY TYPE III)
This syndrome was first described by Saethre in 1931 and by Chotzen in 1932. The predominant features include a brachycephalic skull, a low-set frontal hairline, prominent crus helicis extending through the conchal bowl, facial asymmetry, and ptosis of the eyelids (Figure 23.5). The mode of inheritance is autosomal dominant, with wide variability in expression. The diagnosis is confirmed by identification of a mutation in the TWIST-1 gene on chromosome 7p21, which is believed to result in a dysregulation between bone deposition and maintenance of suture patency.
The craniofacial features include unicoronal or bicoronal synostosis, which is often asymmetric giving a plagiocephalic appearance and contributes to facial asymmetry. The low-set hairline is also a constant feature of this syndrome. The facial asymmetry is often accompanied by deviation of the nasal septum and maxillary hypoplasia with a narrow palate. Intelligence is usually normal. A partial syndactyly involving the index and long fingers is often observed, and short stature is also a frequent finding.
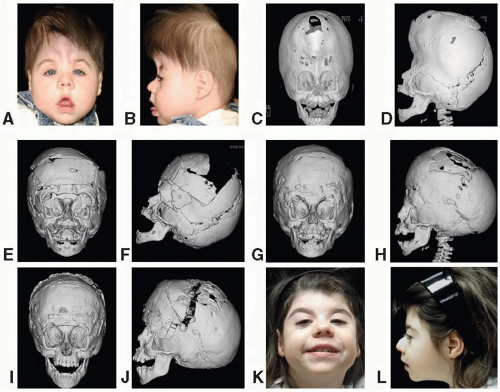 FIGURE 23.5. Saethre-Chotzen syndrome. Note the brachycephalic and turricephalic skull, low-set frontal hairline, and ptosis of the eyelids (A–D). In this case, the patient had multiple-suture craniosynostosis necessitating a strip craniectomy in early infancy. This was followed by fronto-orbital advancement at 10 months to improve brow position and frontal bone contour (E, F), followed by posterior cranial vault distraction at age 24 months to remove occipital flatness and the increased height of the posterior skull while further expanding the intracranial volume (G, H). With a decreased posterior vertical height, the increased anterior vertical height and recurrent brow retrusion are addressed during a second FOA (I–L). |
Patients with Saethre-Chotzen have a high incidence of need for reoperation after cranial vault expansion, ranging from 42% to 65%. Several reports have demonstrated high reoperation rates for poor growth after fronto-orbital advancement (FOA); however, a recent report demonstrated that this patient population also carries a greater than 40% risk of developing elevated ICP after the initial cranial vault expansion. Clearly, such a high-risk population warrants strict monitoring and their families should be made aware that the majority of these patients require more than one cranial vault procedure in the course of their treatment.
MUENKE SYNDROME
Unlike other eponymous craniosynostosis syndromes, Muenke syndrome derives its name from the first report of the genetic mutation rather than the phenotype. The mutation is a pro-250Arg mutation in FGFR-3 on chromosome 4p, which has an incidence of 1 in 10,000 and demonstrates an autosomal dominant inheritance pattern with variable expressivity. It is estimated that Muenke syndrome may be present in 10% of unicoronal or bicoronal synostosis cases that were previously believed to be non-syndromic in origin. The most consistent features include craniosynostosis of the coronal sutures, hearing loss, developmental delay, and thimble-like middle phalanges. Midface hypoplasia is not a common finding. Muenke syndrome exhibits significant variability in the presentation of craniosynostosis between genders, where 88% of females and 76% of males with the mutation have craniosynostosis. While bicoronal synostosis is the most common presentation for both sexes, males demonstrate a much higher incidence of unicoronal craniosynostosis (37% bicoronal vs. 29% unicoronal) than females (58% bicoronal vs. 20% unicoronal). The pattern of sensorineural hearing loss found in these patients is characteristically a bilateral, symmetric, low- to mid-frequency pattern.
The clinical relevance of Muenke syndrome lies in the course of these patients after their initial surgical treatment. In a large retrospective review for patients with coronal craniosynostosis, the reoperation rate for elevated ICP in Muenke syndrome was five times more common than in those without the mutation. Others have also found rates of reoperation to be much higher and aesthetic outcomes to be poorer in Muenke syndrome. In a patient population that is already at an increased risk for developmental delays, and lack significant extracranial signs of their genetic diagnosis, a high level of suspicion and low threshold for genetic testing must be had when evaluating patients with unicoronal or bicoronal synostosis, particularly those with a family history.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


