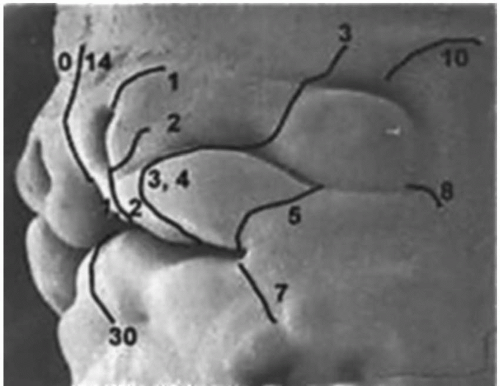
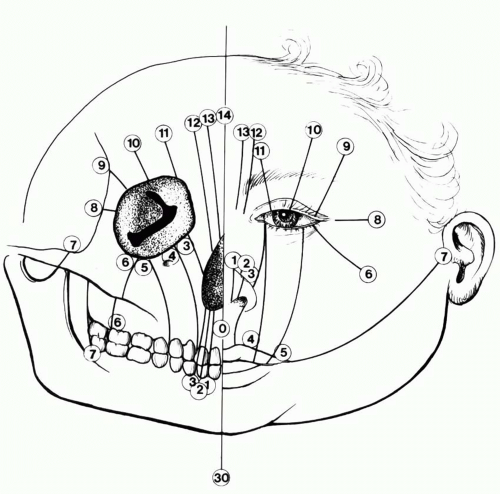
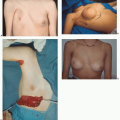
Number 0 Cleft
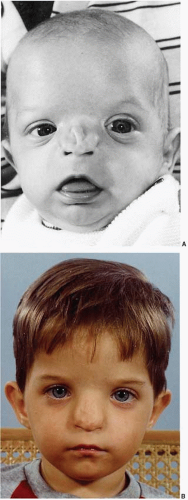
The number 0 cleft has been called median craniofacial dysraphia, centrofacial microsomia, frontonasal dysplasia, or median cleft face syndrome—but for accuracy it is the facial manifestation or lower half of “median craniofacial dysplasia.” Patients with this midline facial cleft may have a cranial extension or a number 14 cleft. The number 0 Tessier craniofacial clefts are unique in that there may be deficient, normal, or excess tissue. Tissue agenesis and holoprosencephaly (the hypoplasias) are one end of the spectrum, and frontonasal hyperplasia and excessive tissue (the hyperplasias) are the other end. Median anomalies with normal tissue volume occupy the middle portion of the spectrum (
Table 26.1).
5Median craniofacial hypoplasia (deficiency of midline structures): A deficiency may manifest as hypoplasia or agenesis in which portions of midline facial structures are missing. This developmental arrest may range from the mildest form of hypoplasia of the nasomaxillary region and hypotelorism to a severe form of cyclopia, ethmocephaly, or cebocephaly. The subcategories in
Table 26.1 demonstrate that the severity of the facial anomalies generally correlate with the severity of brain abnormality and mental retardation. Clinically, it may be important to distinguish among patients with poor brain differentiation (alobar holoprosencephaly) who may die in infancy from those with a better prognosis (lobar brain).
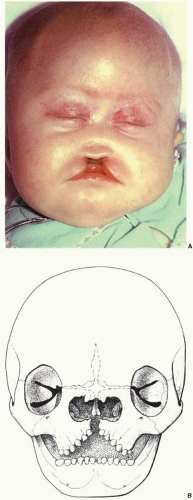
Soft tissue deficiencies with Tessier 0 clefts include the upper lip and nose. Agenesis or hypoplasia may result in a false median cleft lip and absence of philtral columns. When a wide central cleft exists, it typically extends the length of the upper lip and into the nasal floor (
Figure 26.3A). With nasal anomalies the columella may be narrowed or totally absent. The nasal tip may be depressed from lack of septal support. The septum may often be vestigial with no caudal attachment to the palate. Dental abnormalities may include a single maxillary central incisor or even absent central maxillary incisors.
Skeletal deficiencies range from separation between the upper central canines to absence of the premaxilla and a cleft of the secondary palate (
Figure 26.3B). Nasal deficiency may include partial or total absence of the septal cartilage and even nasal bones. The bone defect may extend cephalad into the area of the ethmoid sinuses and result in hypotelorism or cyclopia.
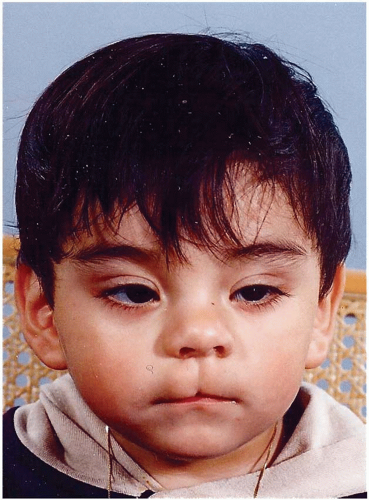
Median craniofacial dysraphia (normal tissue volume but clefted): These Tessier 0 clefts have normal tissue volume but are abnormally split (true median cleft lip) or displaced (encephalocele).
Soft tissue involvement: When an isolated cleft of the upper lip is not associated with tissue deficiency (e.g., absent nasal septum) or tissue excess (e.g., duplicated septum), it is considered a “true” median cleft lip (
Figure 26.4). With a true median cleft lip there is a split between the median globular processes; whereas, with a false median cleft lip an agenesis of the globular processes may occur.
Skeletal involvement: When the true median cleft passes between the central incisors, the cleft can continue posteriorly as a midline cleft palate. When the cleft encroaches into the interorbital region, hypertelorbitism may occur.
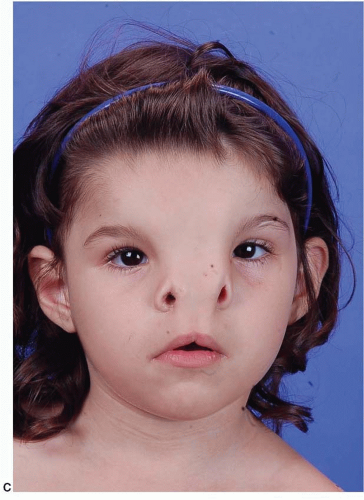
Median craniofacial hyperplasia (excess of midline tissue): This spectrum of midline anomalies includes all forms of excess tissue from a thickened or duplicated nasal septum to the more severe forms of frontonasal dysplasia (
Figure 26.5).
Soft tissue midline excess may be manifested in the lip with broad philtral columns or a duplication of the labial frenulum. The nose may be bifid with a broad columella and mid-dorsal furrow. The alar and upper lateral cartilages may be displaced laterally.
Skeletal excess in a wide 0 facial cleft can be seen as a diastema between the upper central incisors. A duplicate nasal spine may exist. A keel-shaped maxillary alveolus with anterior teeth angled toward the midline creating an anterior open bite is characteristic. Central midface height is shortened. The cartilaginous and bony nasal septum is thickened or duplicated. The nasal bones and nasal process of the maxilla are broad, flattened, and displaced laterally from the midline. Ethmoidal and sphenoidal sinuses may be enlarged, contributing to symmetrical widening of the anterior cranial fossa and hypertelorism.
Number 1 Cleft
Soft tissue involvement: The number 1 cleft, similar to the common cleft lip, passes through the cupid’s bow and then the alar cartilage dome. Notching in the area of the soft triangle of the nose is a distinct feature (
Figure 26.6A). The columella may be short and broad. The nasal tip and nasal septum deviate away from the cleft. When the cleft is evident medially to a malpositioned medial canthus, telecanthus may result. With accompanying cranial extension as a number 13 cleft, vertical dystopia may be present.
Skeletal involvement: An alveolar cleft would pass between the central and lateral incisors (
Figure 26.6B). This paramedian cleft separates the nasal floor at the pyriform aperture just lateral to the nasal spine. The cleft may extend posteriorly as a complete cleft of the hard and soft palate. Extension of the cleft in a cephalad direction is through the junction of the nasal bone and the frontal process of the maxilla.