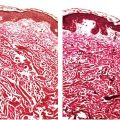
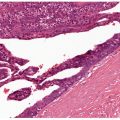
Figure 10-1 Discoid lupus erythematosus. A: An adult with discoid plaques of lupus erythematosus on the upper back: Areas of erythema show stromal inflammation, and paler zones demonstrate scarring. B: The epidermis has lost its rete ridge pattern and shows follicular plugging. There is a brisk mononuclear inflammatory infiltrate near the dermal–epidermal junction and around appendages. The faint bluish background is mucin among collagen bundles. C: Focal subepidermal vacuolization and lymphoplasmacytic infiltrates approximating squamatized basilar keratinocytes. Thickening of the basement membrane is noticeable around follicular ostia. D: Lymphoplasmacytic infiltrates surround eccrine coils in the deep dermis. Interstitial mucin deposits are present in the adjacent stroma. E: Combination Alcian blue–periodic acid–Schiff stain demonstrating a thickened and tortuous basement membrane zone and abundant interstitial mucin deposits among collagen bundles.
In many instances, the discoid cutaneous lesions are limited to the face, where the malar areas and the nose are predominantly affected. In addition, the scalp, ears, oral mucosa, and vermilion border of the lips may be involved. In patients with discoid lupus limited to the head and neck, conversion to SLE is rare (5% to 10%) (11).
In patients with disseminated DLE, discoid lesions are seen on the upper trunk and upper limbs, usually, but not always, in association with lesions on the head (12). SLE may eventually develop in up to 20% of these patients (13). Although discoid cutaneous lesions are typical of patients with chronic cutaneous LE, they are also seen in 14% of the patients with SLE (14).
Histopathology. In most instances of discoid lesions, a diagnosis of LE is possible on the basis of a combination of histologic findings (Fig. 10-1B–E). Changes may be apparent at all levels of the skin, but all need not be present in every case. The findings are summarized as follows:
1. Stratum corneum: hyperkeratosis with follicular plugging
2. Epithelium: thinning and flattening of the stratum malpighii, hydropic degeneration of basal cells, dyskeratosis, and squamatization of basilar keratinocytes
3. Basement membrane: thickening and tortuosity, correlating with deposition of immune reactants (Fig. 10-2)
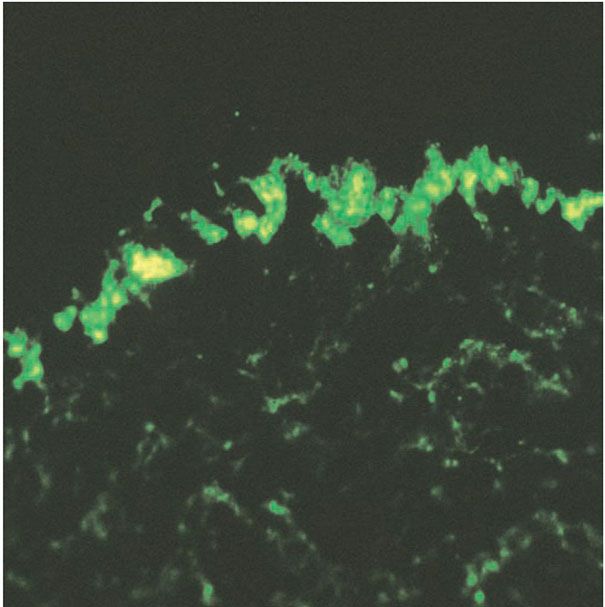
Figure 10-2 Positive lupus band test. Granular deposits of immunoglobulin G in the basement membrane zone on direct immunofluorescence of lesional skin.
4. Stroma: a predominantly lymphoplasmacytic infiltrate arranged along the dermal-epidermal junction, around hair follicles and eccrine glands, and in an interstitial pattern; interstitial mucin deposition; edema, vasodilation, slight extravasation of erythrocytes
5. Subcutaneous: possible slight extension of the inflammatory infiltrate
The stratum corneum is usually hyperkeratotic. Parakeratosis is not conspicuous, and it may be absent. Keratotic plugs are found mainly in dilated follicular openings (Fig. 10-1B, C), but they may occur in the openings of eccrine ducts as well. Follicular channels in the dermis may contain concentric layers of keratin instead of hairs.
The epidermal changes vary with the clinical character of lesions; there may be thinning and flattening of the stratum malpighii. A clinically verrucous lesion shows a hyperplastic epidermis with hyperkeratotic scale that simulates a hypertrophic actinic keratosis or even a superficially invasive squamous cell carcinoma (Fig. 10-3A, B) (15).

Figure 10-3 Hypertrophic or verrucous lupus erythematosus. A: A woman with hyperkeratotic scaly plaques on an erythematous base in a photo distribution. B: A hyperplastic epidermis with hypergranulosis and a brisk lichenoid mononuclear infiltrate, histologically mimicking lichen planus.
The most significant histologic change in LE is hydropic degeneration of the basal layer, also referred to as liquefactive or vacuolar degeneration. This change is characterized by vacuolar spaces beneath and between basilar keratinocytes (Fig. 10-1C). In its absence, a histologic diagnosis of LE should be made with caution and only when other histologic findings greatly favor such a diagnosis. In addition to liquefactive degeneration, basilar keratinocytes may show individual cell necrosis (apoptosis) and acquire elongated contours similar to their superficial counterparts rather than retain their normal columnar appearance (squamatization). Frequently, the undulating rete ridge pattern is lost and is replaced by a linear array of squamatized keratinocytes (Fig. 10-1C) (8).
The basement membrane, normally delicate and inconspicuous, appears thickened and tortuous in longstanding lesions (Fig. 10-1E). This change becomes more apparent with periodic acid–Schiff (PAS) stains, and may be found along the follicular-dermal junctions as well. These findings correlate with locations of immunoreactant deposits found on direct immunofluorescence testing of affected skin (Fig. 10-2). By contrast, in areas of pronounced hydropic degeneration of the basal cells, the PAS-positive subepidermal basement zone may be fragmented and even absent (16). Capillary walls may also show thickening, homogenization, and an increase in the intensity of the PAS reaction.
The inflammatory infiltrate in the dermis is usually lymphocytic admixed with plasma cells (Fig. 10-1C, D). Its distribution is a clue to the diagnosis of LE. In active lesions, the infiltrate can be found approximating the dermal–epidermal junction associated with hydropic degeneration. In hair-bearing areas, the infiltrate is located around folliculosebaceous units and eccrine glands (Fig. 10-1C). Frequently, hydropic changes in the basal layer of the hair follicles may be seen. This may be of diagnostic value in the absence of dermal–epidermal changes. Ongoing inflammation results in atrophy and eventual dropout of the pilosebaceous units. A patchy inflammatory infiltrate may also be present in the upper dermis in an interstitial pattern and around eccrine coils. Occasionally, the infiltrate extends into the subcutaneous fat.
The dermis may show edema, ectatic vessels, and foci of hemorrhage. In dark-skinned persons, melanophages are commonly seen. Increased amounts of ground substance (hyaluronic acid) are common in the middle and lower dermis and are best demonstrated with colloidal iron or Alcian blue stains (Fig. 10-1E) (17). Fibrinoid deposits in the dermis are encountered only rarely in discoid lesions.
Colloid bodies, referred to in lichen planus as Civatte bodies, are apoptotic keratinocytes that present as round-to-ovoid, homogeneous, eosinophilic structures. They may be seen in lesions of DLE and also in other inflammatory processes where there is damage to basilar keratinocytes (poikiloderma, lichen planus, fixed drug eruption, and lichenoid keratoses). They measure approximately 10 μm in diameter and are present in the lower epidermis or in the papillary dermis. When located in the dermis, colloid bodies are PAS positive and diastase resistant and, on direct immunofluorescence staining, are often found to contain immunoglobulins (Igs) (IgG, IgM, IgA), complement, and fibrin. This staining does not represent an immunologic phenomenon but is the result of passive adsorption.
Pathogenesis. Refer to the end of discussion of SLE.
Differential Diagnosis. The epidermal changes seen in DLE must be differentiated from lichen planus since both diseases may show hydropic degeneration of the basal cell layer. In lichen planus, there is wedge-shaped hypergranulosis and triangular elongation of rete ridges described as “saw-toothing,” which are not observed in DLE; in DLE, the rete ridge pattern frequently appears flat. In addition, lichen planus shows a superficial infiltrate (not superficial and deep) and lacks plasma cells and stromal mucin. (For a discussion of the overlap of lichen planus with lupus erythematosus, see Chapter 7.)
Patchy dermal lymphocytic infiltrates may be seen in five disorders that begin with the letter L (called the five Ls). They are LE, lymphocytic lymphoma, lymphocytoma cutis, polymorphous light eruption of the plaque type, and lymphocytic infiltration of the skin of Jessner. Some may also add lues (syphilis) and Lyme disease.
In the absence of significant subepidermal vacuolization, LE must be differentiated from the other six diseases:
• In lymphocytic lymphoma, atypical lymphocytes are present, are tightly packed, have an interstitial distribution (“Indian filing”), and do not surround pilosebaceous units as in LE.
• In lymphocytoma cutis (see Chapter 31), the infiltrate usually is heavier than in LE, may have an interstitial component, shows no tendency to arrange itself around pilosebaceous structures, and often contains an admixture of larger, paler lymphocytes arranged in lymphoid follicles, mimicking germinal center formation.
• In the plaque type of polymorphous light eruption, there is often a prominent band of papillary dermal edema. The infiltrate is more intense in the superficial dermis than in deep dermis and is occasionally admixed with neutrophils. It does not have a folliculocentric arrangement and is not usually accompanied by stromal mucin deposition (see Chapter 12).
• In Jessner lymphocytic infiltration of the skin, the dermal infiltrate may be indistinguishable from that seen in early, nonscarring, or purely dermal lesions of LE; however, plasma cells and mucin are absent. The presence of increased numbers of B lymphocytes in the infiltrate may also help distinguish this from LE (see later text) (18).
• In lues, lichenoid lymphoplasmacytic infiltrates are not associated with mucin deposits. Confirmatory antitreponemal antibody immunohistochemical stains or silver stains, such as a Steiner stain, are positive in lues and negative in lupus.
• In Lyme disease, there are lymphoplasmacytic infiltrates around dermal vessels. Silver and immunohistochemical stains should reveal spirochetal organisms, although they are sparse and difficult to detect.
As will be discussed in later sections, particular forms of LE may be associated with certain drug exposures. 5-Fluorouracil (5-FU) and capecitabine (a pro-drug of 5-FU) have been associated with the induction of DLE-like lesions. Other reported causes of drug-induced discoid lupus include uracil-tegafur (UTF) and infliximab.
Principles of Management. Refer to the end of discussion of SLE.
Verrucous Lupus Erythematosus
Clinical Summary. An exaggerated proliferative epithelial response may occur in approximately 2% of patients with chronic cutaneous LE. These are manifest as verrucous-appearing lesions (Fig. 10-3A). Clinically, two types of lesions have been reported in this subset of LE. Lesions may simulate hypertrophic lichen planus or keratoacanthomas. They occur on the face (nose, chin, and lips), arms, dorsal aspects of the hands, and occasionally the back. The presence of typical DLE lesions elsewhere is a helpful clue to the diagnosis.
Histopathology. Histologically, the epidermis is papillomatous and hyperplastic, and is surmounted by hyperkeratotic scale. Large numbers of dyskeratotic keratinocytes are seen in the lower portion of the epithelium, associated with a bandlike mononuclear infiltrate (Fig. 10-3B). Older lesions show a thickened basement membrane zone. A second pattern consists of a cup-shaped, keratin-filled crater surrounded by an acanthotic epidermis with elongated rete ridges and a sparse mononuclear infiltrate. These changes, in the presence of a deep dermal perivascular, periappendageal, and interstitial infiltrate and mucin deposition, suggest a diagnosis of verrucous LE. CD123, a marker for plasmacytoid dendritic cells, typically marks large numbers of mononuclear cells in LE. The marker has been reported to be of utility in distinguishing hypertrophic LE from squamous cell carcinomas (19).
Pathogenesis. Refer to the end of discussion of SLE.
Differential Diagnosis. Histologically, lesions may simulate hypertrophic lichen planus, keratoacanthomas, and squamous cell carcinomas. Careful examination of the basement membrane zone for thickening and tortuosity, and stromal mucin deposition helps distinguish these entities.
Principles of Management. Refer to the end of discussion of SLE.
Tumid Lupus Erythematosus
Clinical Summary. The dermal form of LE without epithelial changes is known as tumid LE or lupus erythematosus tumidus (LET). Clinically, affected patients display indurated urticarial papules, plaques, and nodules without erythema, atrophy, or ulceration (Fig. 10-4A).
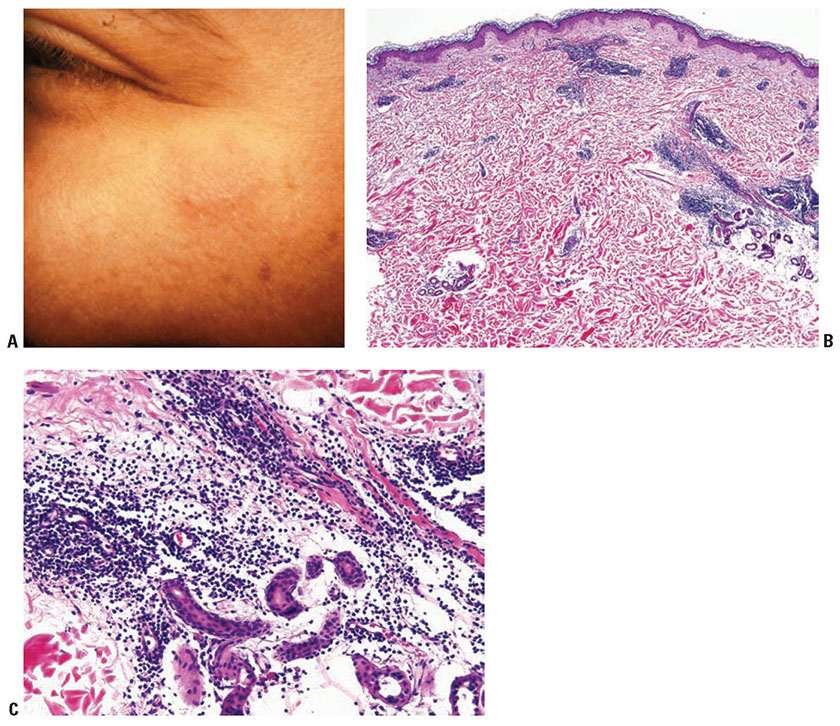
Figure 10-4 Tumid lupus erythematosus. A: A barely raised, slightly hyperpigmented plaque on the cheek, without significant scaling, erythema, or atrophy. B: An angiocentric and periappendageal dermal inflammatory infiltrate. The dermal–epidermal junction of this specimen fails to show interface alterations. C: A lymphocytic infiltrate admixed with plasma cells surrounds and separates eccrine glands and adjacent collagen bundles. The faint bluish hue of the matrix is indicative of mucin deposition.
Histopathology. Histologically, superficial and deep dermal perivascular, interstitial, and periappendageal lymphocytic infiltrates associated with stromal mucin deposits are observed (Fig. 10-4B, C) (20). Unlike discoid lupus, in which discoid lesions may coexist with SLE, the diagnosis of LET typically excludes the presence of SLE (21). Changes at the dermal–epidermal junction, such as liquefaction degeneration or basement membrane thickening are not seen.
Pathogenesis. Refer to the end of discussion of SLE.
Differential Diagnosis. As described later, there is significant overlap of LET with other entities such as lymphocytic infiltrate of Jessner (22).
Principles of Management. Refer to the end of discussion of SLE.
Lupus Erythematosus Profundus/Panniculitis
Clinical Summary. LE may show changes in adipose tissue associated with the chronic cutaneous or systemic forms in 1% to 3% of patients. Two thirds of affected patients have DLE lesions, which may be present either directly overlying the area affected by deeper inflammation or remote from it. A minority of patients have only panniculitis without other cutaneous involvement. Women are affected three to four times more often than men. Typically, multiple discrete, firm, deep nodules arise on the face, arms (particularly the deltoid area), chest, and/or buttocks. The legs and back may be affected as well. The overlying skin may be normal, erythematous, or atrophic. The panniculitis resolves, leaving depressed atrophic scars due to loss of fat with minimal epidermal change.
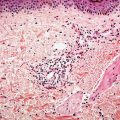
Histopathology. Subcutaneous adipose tissue may be involved with or without inflammation in the dermis or dermal–epidermal junction. Salient histologic findings include a predominantly lobular panniculitis with lymphocytic infiltrates and plasma cells, occasionally forming germinal centers. Although typically described as a lobular panniculitis, septal involvement is reported in 82% of patients in one study (23). Vascular changes include endothelial prominence, thrombosis, calcification, or perivascular fibrosis (“onion-skin” appearance). Fat necrosis occurs with fibrin deposition and lymphocytic nuclear dust which eventuates into hyalinization of adipose lobules (Fig. 10-5A, B). Stromal mucin deposition is conspicuous in well-established lesions. The intensity of the infiltrates lessens over time as the hyalinization progresses.
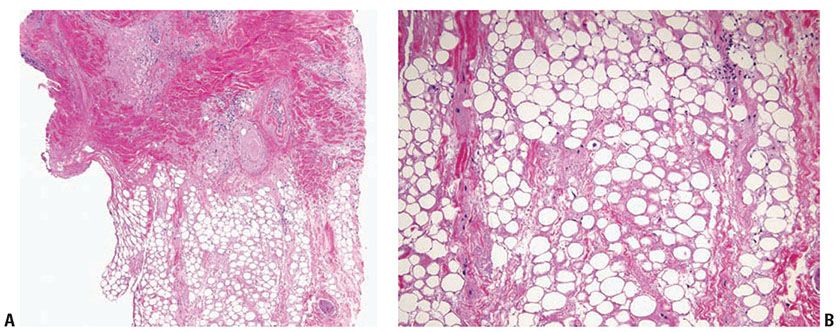
Figure 10-5 Lupus panniculitis. A: A perivascular and perifollicular inflammatory infiltrate is present in the deep dermis and extends into adipose tissue in an interstitial pattern. B: Adipocytes vary in size and are embedded in a hyalinized matrix. Longstanding lesions show minimal inflammation and prominent hyalinization.
Pathogenesis. Refer to the end of discussion of SLE.
Differential Diagnosis. Some observers believe that LE profundus may represent a subcuticular T-cell dyscrasia related to CTD, which has an indolent biological behavior (24). Other feel that lupus panniculitis may be differentiated from subcutaneous panniculitis-like T-cell lymphoma (SPTCL) by histologic features: the absence of other features of LE and the presence of a monoclonal, predominantly α/β CD8 cytotoxic phenotype in atypical lymphocytes favors SPTCL (23).
Principles of Management. Refer to the end of discussion of SLE.
Subacute Cutaneous Lupus Erythematosus
Clinical Summary. Subacute cutaneous lupus erythematosus (SCLE), described in 1979, represents about 9% of all cases of lupus erythematosus. It is characterized by extensive, erythematous, symmetric nonscarring and nonatrophic lesions that arise abruptly on the upper trunk, extensor surfaces of the arms, and dorsa of the hands and fingers. This eruption has two clinical variants: (a) papulosquamous lesions and (b) annular to polycyclic lesions (Fig. 10-6A). Frequently both types of lesions are seen. In some instances, vesicular and discoid lesions with scarring may coexist.
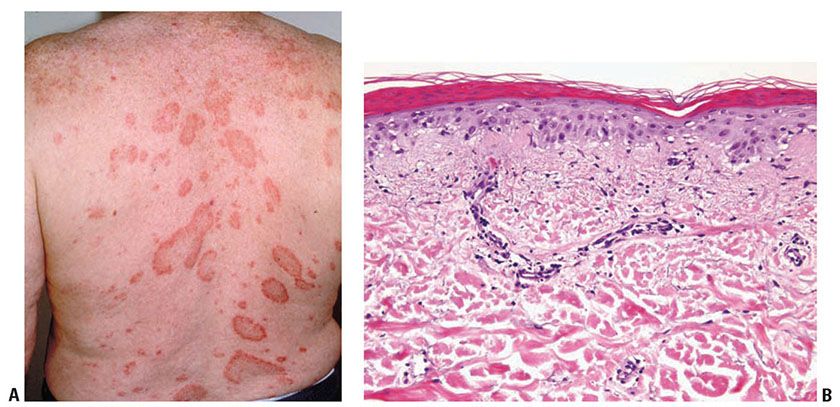
Figure 10-6 Subacute lupus erythematosus. A: Variably sized annular scaly patches on the upper back. Scarring and atrophy, as commonly seen in discoid/chronic cutaneous lupus, are uncommon. B: A sparse mononuclear inflammatory infiltrate in the upper dermis associated with pronounced dermal edema. There is continuous subepidermal vacuolization, focal hemorrhage, and slight mucin deposition.
Patients with SCLE may have mild systemic involvement, particularly arthralgias. Approximately 50% fulfill at least four of the criteria for SLE. Conversely, 10% to 15% of SLE patients exhibit cutaneous lesions of SCLE (25). Severe SLE, with renal or cerebrovascular disease, develops in only 10% of SCLE cases (26,27). Serologic studies show 70% of affected patients to have the anti-Ro (SSA) antibody. Patients with SCLE often bear the HLA-DR2 and HLA-DR3 phenotype. SCLE may occur asynchronously with other CTDs such as Sjögren syndrome and morphea.
Histopathology. See the histopathology section of neonatal LE and Fig. 10-6B.
Pathogenesis. Refer to the end of discussion of SLE.
Differential Diagnosis. Subacute LE is a commonly reported phenotype in drug-induced lupus erythematosus (DILE). About 25 to 30% of cases of SCLE may be attributable to medications, and numerous medications have been reported to cause DILE with a subacute morphology (28). Distinguishing a medication induced reaction can be difficult, though systemic symptoms are typically absent in DILE as opposed to SCLE. The most common medications implicated include hydrochlorothiazide, ACE inhibitors, Calcium channel blockers, tumor necrosis factor (TNF) inhibitors, terbinafine, and chemotherapeutic agents (29), though the list is long and ever-expanding (28).
Principles of Management. If the onset of the eruption coincides with the administration of a new medication, particularly one on the above list, discontinuation may permit resolution of the eruption. Notably, some patients may have “drug-unmasked” lupus rather than pure “drug-induced” lupus. If the eruption persists, sunscreens and therapeutic agents as discussed at the end of the section on systemic lupus are used.
Neonatal Lupus Erythematosus
Clinical Summary. Neonatal LE has clinical and histologic skin changes and serologic findings similar to those of SCLE. Children of mothers with active SLE or Sjögren syndrome may develop LE-like symptoms in the neonatal period. Anti-Ro/SSA is the predominant autoantibody and is found in approximately 95% of cases. This may result in a transient syndrome characterized by widespread erythematous desquamate polycyclic, annular, usually nonscarring lesions. Atrophy may be present in some cases. There is typically central facial involvement. Less common cutaneous manifestations include urticarial lesions (30). There is associated photosensitivity, transient thrombocytopenia, mild hemolytic anemia, leukopenia, and congenital heart block.
Histopathology of SCLE and Neonatal LE. Histologic changes differ in degree from those in the discoid lesions and are most intense at the dermal–epidermal interface (Fig 10-6B). They consist of the following:
1. Hydropic degeneration of the basilar epithelial layer, sometimes severe enough to form clefts and subepidermal vesicles
2. Colloid bodies in the lower epidermis and papillary dermis (common)
3. Edema of the dermis, which is more pronounced than in discoid lesions
4. Focal extravasation of erythrocytes and dermal fibrinoid deposits (common)
5. Less prominent hyperkeratosis and inflammatory infiltrate than in discoid lesions (31)
Pathogenesis. Neonatal LE is related to passage of maternal IgG antinuclear antibodies (particularly anti-Ro/SSA, anti-La/SSB, or anti-U1RNP autoantibodies) through the placenta. These changes have their onset at birth to 2 months and usually resolve in the first 6 to 9 months of life with decreasing levels of maternal antibodies. Heart block occurs in approximately 50% of affected infants, usually without associated skin lesions, and may be fatal. Of interest, individuals affected with transient neonatal LE may develop SLE as young adults (20). Animal model studies suggest that reactivity to the p200 region of the Ro52 protein and antibodies targeting calcium channel blockers may have a role in cardiac involvement in affected children (32). Of interest, multigenerational genetic studies suggest that mothers of neonates with lupus accumulate genetic risk preferentially from neonatal lupus children’s grandparents (32).
Differential Diagnosis. The eruption in the clinical setting of a neonate born to a mother with LE and interface changes on biopsy is characteristic.
Principles of Management. Treatment includes management of the cutaneous eruption as well as cardiomyopathy. All children with suspected neonatal lupus require immediate evaluation of the cardiac conduction system. Parents of children with neonatal lupus should be evaluated, as this may be the presenting sign of previously subclinical disease. Fluorinated steroids, intravenous immunoglobulin, and hydroxychloroquine are used in the prevention and treatment of the disease.
Systemic Lupus Erythematosus
Clinical Summary. In SLE, the cutaneous manifestations usually appear less suddenly than in SCLE and are less pronounced, such that the signs and symptoms of systemic disease usually overshadow the often subtle form of skin involvement. Usually systemic manifestations, especially arthritis, precede the cutaneous lesions. Only 20% of SLE patients demonstrate prominent cutaneous features at the onset of their disease, but approximately 80% will exhibit cutaneous lesions in the course of their disease (33,34).
The cutaneous manifestations consist of malar erythema, photosensitivity, palmar erythema, periungual telangiectases, diffuse hair loss as a result of telogen effluvium, and urticarial vasculitis or bullous lesions. The erythematous lesions of SLE consist of erythematous, slightly edematous patches without scaling or atrophy. As a rule, the patches are ill-defined. The most common site of involvement is the malar region, but any area of the skin may be involved, particularly the palms and fingers. Occasionally, lesions show a petechial, vesicular, or ulcerative component. Senescent lesions may assume the appearance of poikiloderma atrophicans vasculare.
Well-defined “discoid” lesions with atrophic scarring, as seen typically in DLE, occur in about 15% of the patients with SLE. They may precede all other clinical manifestations of SLE. A relatively benign course characterizes SLE in most patients with preceding DLE (35); however, many patients usually have had persistent multiple abnormal laboratory findings from the beginning. This is in contrast to cases of simple DLE, in which most abnormal laboratory findings are transient.
Two variants of SLE—SLE with genetic deficiency of complement components and bullous SLE—bear mention. In the former, the onset of SLE occurs in early childhood and is transmitted in an autosomal recessive fashion and often affects several siblings (36). Deficiencies of C2 and C4 result in extensive lesions similar to DLE, with scaling, atrophy, and scarring associated with sensitivity to sunlight. There can be associated central nervous system involvement and glomerulonephritis, which may be fatal (37).
In bullous SLE, subepidermal blisters may arise in previously involved or uninvolved areas. They may form large hemorrhagic bullae or herpetiform vesicles, which arise suddenly, and are clinically similar to lesions of bullous pemphigoid or dermatitis herpetiformis.
Rowell syndrome is a distinct clinical presentation of lupus with erythema multiforme–like lesions. Patients have speckled antinuclear and anti-La antibodies and test positive for rheumatoid factor (38).
The coexistence of SLE and diffuse systemic sclerosis (scleroderma) or dermatomyositis has been repeatedly described. It is known as overlap syndrome and refers to the coexistence of two related but separate diseases. Similarly, there are reports of systemic lupus and coexistent eosinophilic fasciitis (39,40). Such instances of overlap are in contrast to mixed connective tissue disease (MCTD), which is recognized as a separate disease entity.
Many drugs are associated with induction of LE, the majority of which induce predominantly systemic symptoms such as arthritis, serositis, lymphadenopathy, and fever. A few are associated with specific cutaneous manifestations. They are listed in Table 10-1, and are discussed further in the section on drug-induced lupus erythematosus in Chapter 11.
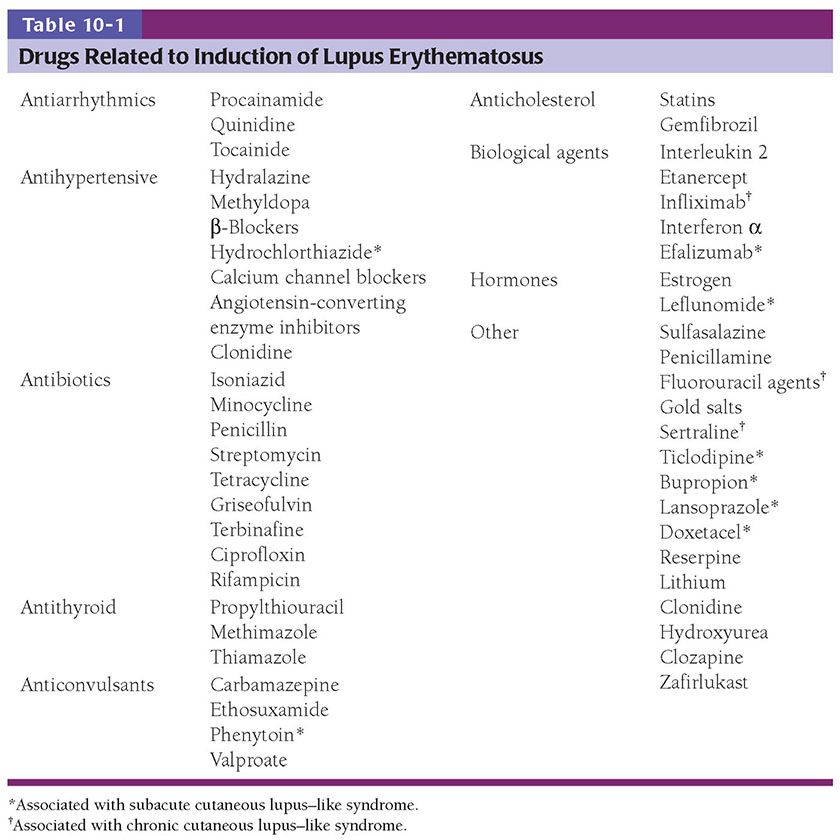
Histopathology. Early lesions of SLE of the erythematous, edematous type may show only mild, nonspecific changes. In well-developed lesions, the histologic changes correspond to those described for SCLE (Fig. 10-6B): Hydropic degeneration of the basal cell layer occurs in association with edema of the upper dermis and extravasation of erythrocytes. Fibrin deposits in the connective tissue of the skin are often seen in erythematous lesions. They appear as granular, strongly eosinophilic, PAS-positive, diastase-resistant deposits between collagen bundles, in the walls of dermal vessels, in the papillary dermis, or beneath the epidermis in the basement membrane zone. Fibrinoid deposits are not specific for LE. They are seen in association with vascular injury, particularly in leukocytoclastic vasculitis.
Subcutaneous fat is often involved in SLE. Changes similar to those in lupus profundus may be seen but are usually milder: There may be focal mucin deposition associated with a predominantly lymphocytic infiltrate. Adipocytes may be separated by edema and fibrinoid deposits. These histologic changes in subcutaneous fat produce no clinically apparent lesions.
Occasionally, palpable purpuric lesions in SLE patients show a leukocytoclastic vasculitis histologically indistinguishable from leukocytoclastic vasculitis of other causes. There is endothelial cell swelling, a neutrophilic inflammatory infiltrate, nuclear dust, perivenular fibrin deposition, and stromal hemorrhage. Urticaria-like lesions may occur and show either a leukocytoclastic vasculitis or a perivascular mononuclear infiltrate not diagnostic of LE (41). In addition, white atrophic lesions may occur in SLE that both clinically and histologically resemble those of malignant atrophic papulosis of Degos (42).
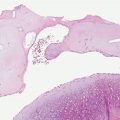
Bullous SLE shows two histologic inflammatory patterns: one neutrophilic and one mononuclear. The neutrophilic type simulates dermatitis herpetiformis or linear IgA disease with the formation of papillary microabscesses (Fig. 10-7A, B) (43). Nuclear dust is seen in the papillary microabscesses and in the upper dermis around and within the walls of blood vessels. Direct immunoelectron studies have localized immunoreactant deposits to beneath the lamina densa; and consist of IgG with or without IgM, and often IgA in a linear or granular pattern. Some patients may also have circulating anti–basement zone antibodies and antibodies directed against type VII collagen. The latter antibodies are similar but not identical to those in epidermolysis bullosa acquisita (44).
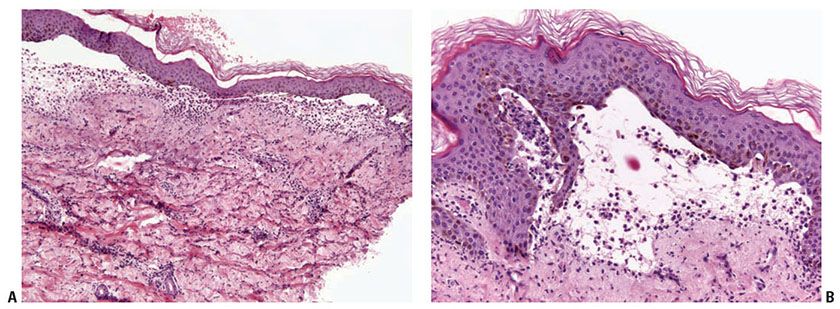
Figure 10-7 Bullous lupus erythematosus, neutrophilic. A: There are broad-based subepidermal blisters associated with marked papillary dermal edema and large numbers of neutrophils. Neutrophils are also present around vessels and among collagen bundles throughout the dermis. Faint interstitial mucin deposits and nuclear dust are present. B: The papillary dermal abscesses simulate findings of dermatitis herpetiformis. The dermal changes as noted in A, however, would not be seen in dermatitis herpetiformis.
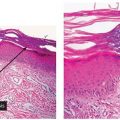
The subepidermal blister associated with a mononuclear inflammatory infiltrate arises in longstanding lesions of cutaneous LE (Fig. 10-8A, B). It likely corresponds to an altered dermal–epidermal interface resulting from inflammation and immune complex deposition. This type of change is a part of the spectrum of LE rather than a distinct entity.
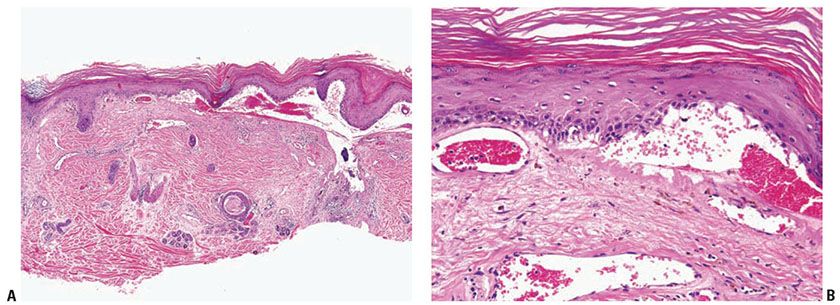
Figure 10-8 Bullous lupus erythematosus, mononuclear. A: A broad subepidermal cleft is present beneath an epidermis that has lost its rete ridge pattern and is surmounted by hyperkeratotic scale. The stroma contains perivascular and periappendageal inflammatory infiltrates. B: Adjacent to the zone of dermal–epidermal separation is a thickened eosinophilic basement membrane zone. Melanophages are scattered in the superficial dermis.
Systemic Lesions. Much of the tissue damage in SLE results from deposition of antibody–antigen complexes in affected organ systems.
A detailed presentation of the arthritic, serosal and cardiac manifestations and classifications of glomerulonephritides is beyond the scope of this section but can be found in the review by Dooley et al. (45).
Antiphospholipid syndrome occurs in patients with SLE and other autoimmune diseases who develop immunoglobulins that can prolong phospholipid-dependent coagulation tests. These immunoglobulins occur in association with SLE and other autoimmune diseases but are found unassociated with them as well. One of these, lupus anticoagulant, occurs in about 10% of SLE patients. Affected patients are at greater risk for thromboembolic disease, including deep venous thrombosis, pulmonary emboli, and other large-vessel thrombosis. Other findings include recurrent fetal wastage, renal vascular thrombosis, thrombosis of dermal vessels (Fig. 10-9A, B), and thrombocytopenia. Anticardiolipin antibody, a second type of antiphospholipid antibody, occurs five times more often than lupus anticoagulant antibody. It is associated with recurrent arterial and venous thrombosis, valvular abnormalities, cerebrovascular thromboses, and essential hypertension (Sneddon syndrome) (46). Other cutaneous findings include livedo reticularis, necrotizing purpura, disseminated intravascular coagulation, and stasis ulcers of the ankles (47).
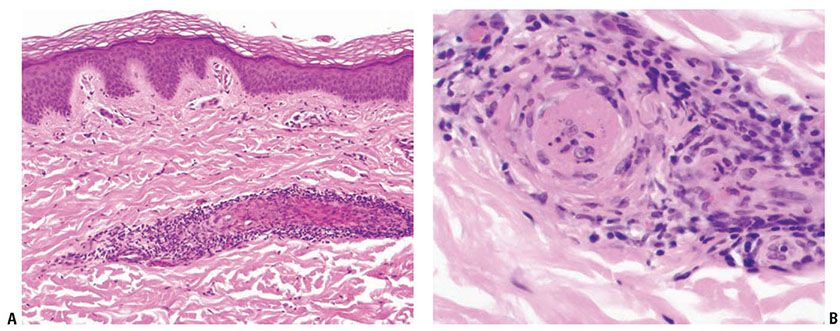
Figure 10-9 Lupus anticoagulant syndrome. A: A superficial dermal vessel is surrounded by lymphocytes. B: High magnification of pale eosinophilic intravascular thrombus.
Pathogenesis. The etiology of LE is considered to be multifactorial (Table 10-2). Studies have demonstrated numerous defects in the innate and adaptive immune system resulting in a widespread loss of self-tolerance. These abnormalities include but are not limited to abnormal responses to ultraviolet radiation, abnormal antigen-presenting-cell function, plasmacytoid dendritic cell function, numerous HLA associations, polymorphisms in TNF-α promoters, abnormal keratinocyte apoptosis, and polymorphisms in the genes coding for interleukin 1 (IL-1) receptor antagonist and IL-10 promoter sequence (48). Aberrant stimulation of intracellular Toll-like receptors (TLRs), specifically TLR7 and TLR9, by endogenous antigens may play a crucial role in the autoimmune feedback loop between T cells and B cells (49). The common final pathway involves the production of autoantibodies that mediate tissue damage (50).

The initial screening test for antinuclear antibodies (ANAs) is indirect immunofluorescence, which provides limited but clinically useful information. Many other assays are available for more specific characterization of ANAs and include immunodiffusion, enzyme-linked immunosorbent assays, immunoprecipitation, and immunoblots.
Indirect immunofluorescence detects nuclear, homogeneous, rim, speckled, and nucleolar patterns. The fluorescence ANA test can be regarded as a specific marker for SLE in a rim pattern with a titer of 1:160 or higher and is indicative of the presence of anti–native or double-stranded (ds) DNA antibodies. Although anti-dsDNA has generally been regarded as the most specific marker of SLE, the lack of sensitivity makes a negative test unhelpful. Other markers that have been examined include antinucleosome antibodies, which may be more prevalent in SLE patients and may develop earlier than anti-dsDNA antibodies. This assay, however, is not widely available and has not been rigorously evaluated (51). At a titer of 1:20, about 20% of patients with DLE, most patients with systemic scleroderma, and as many as 5% of normal persons may have a positive reaction. The presence of anti-Sm is indicative of associated lupus nephritis. Anti-nRNP antibodies are of diagnostic significance only at high titers where they indicate MCTD. ANA-negative sera should be examined for the presence of anti-Ro/SSA and La/SSB antibodies, characteristic of subacute and neonatal LE and SLE with genetic deficiency of complement components.
Direct immunofluorescence studies detect immunoreactant deposits in affected tissues, particularly the skin and kidneys. A skin biopsy (3- to 4-mm punch) is submitted in saline-impregnated gauze or phosphate-buffered saline, snap frozen, sectioned, and incubated with fluorescein-conjugated antisera to IgA, IgM, IgG, and the third component of complement (C3). In a positive test, there is continuous granular deposition of usually two or more immunoreactants in a band along the dermal–epidermal junction (Fig. 10-2). Variables that affect test results are the site of the biopsy (sun exposed vs. sun protected), duration of the lesion (acute, subacute, chronic), and preceding therapy. The frequency and implications of positive results in DLE and SLE are summarized in Table 10-3 (52–54).

Cautious sampling and interpretation of findings are necessary to avoid false-positive and false-negative results. The presence of only one type of immunoreactant, particularly in an intermittent distribution, may be seen in chronically sun-exposed skin or with underlying disorders such as rosacea. False-negative results are often found in acute or subacute lesions and treated lesions. Optimally, an established lesion, present for 3 months or longer, that has not been treated is submitted for study (55).
Differential Diagnosis. Lichenoid eruptions, including lichenoid eruptions related to medications, dermatomyositis and overlap syndromes may show similar histologic features. Correlation with clinical presentation and serologic studies is necessary.
Principles of Management. The treatment modalities for LE cover a wide range of immunosuppressant topical and systemic agents. Typically, the extent of disease dictates the course of therapy. All patients benefit from sun protection with broad spectrum UV blocking chemical and physical agents. Limited cutaneous disease of discoid lupus can be treated with topical mid to high potency steroids. This is often insufficient, however, and systemic therapy with hydroxychloroquine or other antimalarials is necessary; some feel administration of hydroxychloroquine may decrease progression to overt SLE. Patients who fail to respond or with extensive, disfiguring, or symptomatic disease may require more aggressive therapy. These may include methotrexate, thalidomide, prednisone, azathioprine, or mycophenolate mofetil. Of particular importance in discoid lupus is meticulous sun avoidance, which is responsible for many flares in the disease. Subacute cutaneous lupus is similarly treated, though in this instance the strong association with medication-induced disease warrants a thorough review of the patients’ medication list to identify potential triggers. Lupus profundus, due to the subcuticular location of the disease process is not amenable to topical steroids, and patients typically are treated with antimalarials. Systemic lupus is typically treated with systemic immunosuppressants including prednisone, cyclophosphamide, mycophenolate mofetil, methotrexate, and azathioprine. Case reports and small studies have examined the efficacy of thalidomide, high-dose intravenous immunoglobulins, and rituximab or TNF inhibitors, with varying response rates (56). Bullous lupus is often particularly responsive to dapsone and other anti-neutrophilic agents.
Jessner Lymphocytic Infiltration of the Skin
Clinical Summary. Jessner lymphocytic infiltration of the skin, first described in 1953 (57), is not a well-understood entity. It is characterized by asymptomatic papules or well-demarcated, slightly infiltrated red plaques, which may develop central clearing. In contrast to lesions of chronic LE, the surface shows no follicular plugging or atrophy. Lesions arise most often on the face but may also involve the neck and upper trunk (58). Although this disorder has been reported to occur in childhood (59), affected patients are usually middle-aged men and women. Some authors consider this entity to exist on the spectrum of LE and some consider it to be identical to tumid lupus (60,61).
Variable numbers of lesions persist for several months or several years. They may disappear without sequelae or recur at previously involved sites. The eruption may be precipitated or aggravated by sunlight.
Histopathology. The epidermis may be normal or slightly flattened. In the dermis, there are dense perivascular and interstitial infiltrates composed of small, mature lymphocytes admixed with occasional histiocytes and plasma cells (Fig. 10-10A, B). The infiltrate may extend around folliculosebaceous units and into subcutaneous adipose tissue.

Figure 10-10 Jessner lymphocytic infiltrate. A: The epidermis is normal. The dermis contains tightly cuffed lymphocytic infiltrates following blood vessels and focally surrounding appendages. B: Higher magnification shows a purely lymphocytic infiltrate around vascular channels.
Pathogenesis. Opinion varies as to whether Jessner lymphocytic infiltrate of the skin is a distinct entity. The following four views have been expressed: (a) clinical, histologic, and immunohistochemical findings warrant its distinction as a separate entity (62); (b) although some cases represent a distinct entity, others are LE; (c) all cases are LE; or (d) it represents an abortive or initial phase of either LE, plaque type of polymorphous light eruption, lymphocytoma cutis, or lymphocytic lymphoma. In support of the notion that Jessner lymphocytic infiltrate is a form of LE, studies of CD123, a plasmacytoid dendritic cell marker, known to have an effector role in lupus, have shown similar patterns of staining in the inflammatory cells (60). Isolated reports of Jessner lymphocytic infiltrate have been reported in association with angiotensin-converting enzyme inhibitor, enalapril, and a synthetic polypeptide, glatiramer acetate, used to treat multiple sclerosis (63,64). Isolated familial cases have also been reported (65).
Immunohistochemical studies indicate that the predominant cells of lymphocytic infiltrate of the skin are mature T lymphocytes. Phenotyping studies show predominantly CD4+ T helper cells (66). Other studies have suggested a CD8+ cytotoxic phenotype (65). Rearrangement of the T-cell receptor gene has not been detected in the limited number of cases studied. The relative paucity of B lymphocytes may assist in separation of this entity from lymphocytoma cutis, which usually displays larger B-cell components, with or without associated germinal center formation. Lymphoma cutis may be distinguished with cell-marker analysis, which will show a high proportion of B lymphocytes with a less mature phenotype than expected in skin, or cells with aberrant expression of surface antigens.
Differential Diagnosis. The histologic differential diagnosis of lymphocytic infiltration of the skin includes the other six of the seven Ls: LE, polymorphous light eruption, lymphocytoma cutis, lues, Lyme disease, and lymphoma. Jessner lymphocytic infiltrate is limited to sun-exposed skin and lacks the hyperkeratosis, atrophy, interface changes, mucin, and direct immunofluorescence findings noted in lupus. Therefore, lupus should be excluded with serologic and direct immunofluorescence studies. Serologic studies and the clinical distribution of lesions allow separation of lues and Lyme disease from Jessner lymphocytic infiltrates.
Approximately 10% of cases of lupus lack interface alterations and that show negative direct immunofluorescence studies. These cases may initially be placed into the holding category of “lymphocytic infiltrates of the skin.” Polymorphous light eruptions usually show prominent papillary dermal edema; however, plaque-type polymorphous light eruption may histologically overlap with lymphocytic infiltration of the skin. Clinical features may sometimes separate these two entities.
A poorly understood entity histologically similar to Jessner lymphocytic infiltrate is palpable migratory arciform erythema. This entity differs clinically in that it is slightly pruritic and tends to involve the trunk and proximal extremities. It waxes and wanes over days to weeks rather than months. Differences in histology have been reported but have not been validated by multiple studies (67).
Principles of Management. Mid-potency topical steroids are typically utilized for symptomatic management. Sun avoidance is encouraged. In recalcitrant cases, antimalarial drugs and/or corticosteroids are useful in controlling this disorder. Other therapies have included methotrexate, gold, thalidomide, and photodynamic therapy.
MIXED CONNECTIVE TISSUE DISEASE
Clinical Summary. Overlapping findings of SLE, scleroderma (systemic sclerosis), and polymyositis associated with the presence of high titers of anti-U1 ribonucleoprotein (RNP) antibodies have been recognized since 1972 as MCTD (68). The first clue to the diagnosis is usually a positive, high-titer speckled ANA. The clinical presentation includes, but is not limited to, edema of the hands, acrosclerosis, Raynaud phenomenon, synovitis of one or more joints, and myositis that is documented with biopsy or serum elevations of muscle enzymes. Esophageal hypomotility and pulmonary disease may be seen as well. The diagnosis of MCTD may not be made at the outset because symptoms may present in an asynchronous fashion.
Cutaneous LE lesions are found in approximately half of the patients. They cover the entire spectrum of cutaneous LE. Most commonly there are diffuse, nonscarring, poorly demarcated subacute lesions, but some patients show malar eruptions as seen in SLE or persistent scarring lesions of DLE.
Patients with high-titer anti-U1 RNP antibodies have a low prevalence of renal disease and life-threatening neurologic complications. Although the prognosis of MCTD is comparable to that of SLE, death may occur from progressive pulmonary hypertension and cardiac complications (69).
Histopathology. If cutaneous lesions of LE are present, the histologic findings correspond to the type of lesion described in the section on DLE and SLE. MCTD, in contrast with most cases of SLE, has no antibodies to DNA. The absence of such antibodies accounts for the rarity of renal disease.
Indirect immunofluorescence studies show the presence of very high titers of serum antibodies directed against extractable ribonuclease-sensitive antigens known as small nuclear ribonucleoproteins (snRNPs). They characterize MCTD but are not specific for it, and produce a fine speckled ANA pattern at high dilutions. This speckled pattern corresponds to the widespread distribution of snRNP in the nucleus at sites of active gene transcription, where messenger RNA is being processed.
Direct immunofluorescence staining of normal skin shows deposition of IgG in a speckled pattern in epidermal nuclei. Although this finding is typical of MCTD, it is found occasionally also in patients with SLE and other CTDs (70). In addition, patients with MCTD may show a subepidermal lupus band in normal skin. Such a band has been observed in normal sun-exposed skin in about 20% of the cases.
Pathogenesis. The pathogenesis of MCTD is not fully understood, but shows some similarities to LE. Like LE, MCTD is a manifestation of autoimmunity and loss of self-tolerance, driven by aberrations in both innate and adaptive arms of the immune system. Triggering of this process, similar to LE, occurs through interactions of the innate immune system, through TLR with apoptotic cells that have undergone antigen structural modification.
Differential Diagnosis. The differential diagnosis includes many stand-alone diseases that make up the constellation of MCTD. These include polymyositis, SLE, and systemic sclerosis. Although there are distinct serologic patterns that distinguish SLE from MCTD, the underlying immunologic aberration is believed to be similar.
Principles of Management. Similar to other CTDs, the extent of disease dictates the treatment course, and sun protection is essential. For mild disease with arthritic symptoms and cutaneous lesions, nonsteroidal anti-inflammatory agents and topical steroids may be all that is required. Hydroxychloroquine may be used as an adjuvant. For more aggressive disease, systemic steroids and alternate immunosuppressive agents may be required. Pulmonary involvement, in the form of pulmonary hypertension is treated with phosphodiesterase inhibitors and the endothelin receptor antagonist ambrisentan and the synthetic prostaglandin epoprostenol. Phosphodiesterase inhibitors and calcium channel blockers may be used for treatment of Raynaud phenomenon. Intravenous immunoglobulin has been used in recalcitrant cases with success (71).
DERMATOMYOSITIS
Clinical Summary. Dermatomyositis manifests as an inflammatory myopathy with characteristic cutaneous findings. In the absence of cutaneous findings, the diagnosis of polymyositis is applied. Cutaneous findings alone, without muscular involvement, have been termed amyopathic dermatomyositis or dermatomyositis sine myositis (72).
Both dermatomyositis and polymyositis are uncommon diseases that have a similar incidence. Both have two peaks of occurrence: one in childhood and one between the ages of 45 and 65 years (73). The cutaneous eruption and inflammatory myositis may occur asynchronously. Their appearance may be separated by months to years.
The four systemic and single cutaneous diagnostic criteria for dermatomyositis are symmetric proximal muscle weakness, elevated muscle enzymes, an abnormal electromyogram in the absence of neuropathy, consistent muscle biopsy changes, and cutaneous findings (73). Involvement of the skeletal muscles causes progressive weakness, pain, and eventual muscle atrophy. The proximal muscles of the extremities and the anterior neck muscles often are the first to be involved. Involvement of the pharynx may result in dysphagia and aspiration. Involvement of the diaphragm and the intercostal muscles may lead to respiratory failure. Arthritis and arthralgias occur in up to 25% of cases. Interstitial lung disease is becoming increasingly recognized as a frequent occurrence in patients with dermatomyositis (74
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


