Fig. 30.1
Palmoplantar keratoderma in EBS
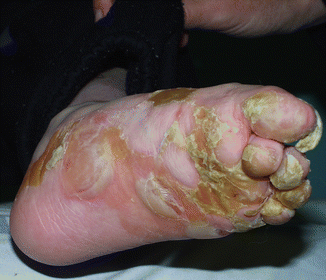
Fig. 30.2
Tense blistering, hyperkeratosis and thickened nails in a boy with EBS gen-se
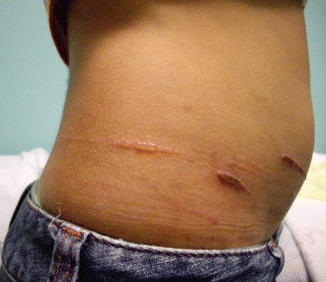
Fig. 30.3
Koebnerization in EBS
In localised EBS, blistering is usually noted after infancy and during early childhood. Skin lesions are localised to areas of mechanical trauma, particularly the hands and feet, and mucosal involvement is rarely noted. Epidermal detachment has been noted to be aggravated by heat and sweating [9]. Some patients have not presented until adulthood when required to do intense physical activity, such as army training (ref). While blistering extent may appear mild to the observer, to the patients this condition may severely limit their quality of life because of its effects on their activities.
At the other end of the clinical spectrum are the EBS subtypes with more generalised blistering. First, the EBS generalised severe subtype is characterised as having extensive and generalised blistering in the neonatal period. These lesions are noted to appear in clusters, hence the characteristic herpetiform feature of this subtype. Mucosal involvement, particularly oral, is a usual finding, and the healing of blisters is usually associated with hyperpigmentation. Patients with the EBS-gen-sev may also present with thickening of their nails, milia formation, atrophic scarring and palmoplantar keratoderma that usually develops after early childhood. In one case report of a patient with EBS-gen-sev, motor delay was also noted, as the toddler could not crawl until the age of 2 years [10]. This is likely owing to the extensive involvement of the skin blisters, which are worsened by mechanical trauma, hence disabling the patient to effectively develop his motor skills. Mortality can be high in the neonatal period if there is extensive blistering, due to infection and septicaemia, and should be managed aggressively as the prognosis is otherwise very good for this form of EBS. Cutaneous lesions of EBS-gen-sev usually improve with age [7].
The second subtype of generalised EBS differs from the EBS-gen-sev subtype in that the blisters, although widespread, are not as severe as the latter. They usually occur along the lines of clothing, in particular the underwear. In this EBS generalised (EBS-gen-intermed) subtype, scarring, milia formation and nail dystrophy are not usually noted. Also, in this subtype, skin lesions are significantly noted to improve with age [10].
EBS with mottled pigmentation (EBS-MP) is a variant of EBS with localised to generalised blistering [11, 12]. Although less common than the three previously mentioned EBS subtypes, the genetic mutations in this subtype are also found in the keratin 5 and keratin 14 genes—more commonly the P25L mutation in keratin 5, although a mutation has also been determined in keratin 14 in one case [12]. EBS-MP presents with localised to generalised blistering at birth. However, the appearance of hyper- and hypopigmented macules in a mottled or mosaic-like arrangement is a characteristic feature of this subtype (Fig 30.4). Blisters have not been noted to always precede the pigmentary changes, unlike post-inflammatory hyperpigmentation commonly seen in other forms of EB. These changes have also been noted to appear in early childhood and fade with age. As age progresses, skin lesions may lessen, although blisters and erosions may still develop periodically. Other features of EBS-MP include EB nevi, nail dystrophy and palmoplantar keratoderma, particularly the punctate type [11, 12].
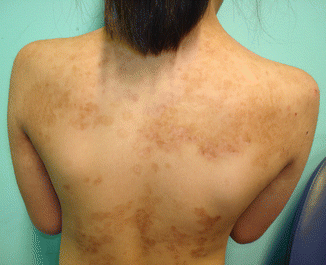
Fig. 30.4
EBS-MP
Another variant of EBS that presents with blistering on the trunk and extremities is EBS with migratory circinate (EBS-migr). This uncommon EBS variant is due to autosomal dominant mutations in the keratin 5 gene. Clinicians may notice a resemblance between EBS-gen-sev and EBS-migr due to the circular arrangement of vesicles in both cases. However, the latter presents with blisters in the characteristic background of prominent circinate and migratory erythema. In this EBS variant, erythematous and annularly arranged blisters appear on the trunk and extremities at birth. Pruritus is usually associated with erupting blisters. These blisters are further aggravated by heat and sweating. Skin lesions heal without scarring, although hyperpigmentation may occur. The nails, eyes, larynx and mucosae are not involved. Milia formation and palmoplantar keratoderma are likewise absent [13].
EBS due to autosomal recessive mutations in keratin 5 and keratin 14 are relatively rare. To date, less than 20 cases of this EBS subtype have been reported in the literature. The clinical spectrum of autosomal recessive EBS (EBS-AR) may range from the mild features of EBS-loc to the more generalised features of EBS-gen-intermed. While some EBS-AR cases report of mild and localised blister formation (Figs 30.5, 30.6, and 30.7), aplasia cutis has been reported on two occasions. Oral, nasal and genital mucosal involvements have likewise been reported. Other clinical features that have been reported in EBS-AR include failure to thrive, iron deficiency anaemia, growth retardation, pruritus and EB nevi. In this EBS subtype, the homozygous genetic defect in keratin 14 leads to the absence of keratin 14. So far, only one case of recessive EBS due to KRT5 has been reported. Therefore, this may justify why clinical features of this subtype may be more severe when compared with the autosomal dominant EBS subtypes [7, 14].
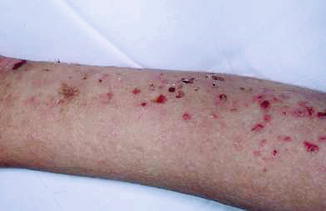
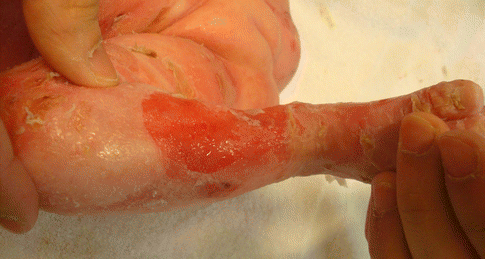
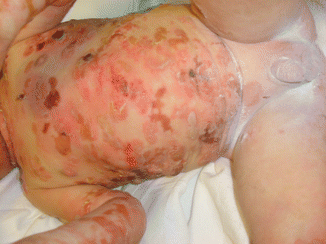
Fig. 30.5
Krt 14 recessive EBS
Fig. 30.6
Krt 5 recessive EBS
Fig. 30.7
Generalized blistering in a baby with EBS-gen-sev due to a keratin 5 mutation.
Recessive EBS may also be due to defects in plakins, namely, plectins and bullous pemphigoid antigen 1 (BPAG1); integrins alpha 6 and beta 4 and exophilin-5 (EXPH5) have also been noted to cause epidermolysis bullosa at the basal level.
The plectin gene (PLEC1) encodes the plectin protein, and mutations in the gene leads to EBS with muscular dystrophy (EBS-MD), EBS with pyloric atresia (EBS-PA) and EBS-Ogna. A defect in PLEC1 that is of autosomal dominant inheritance results in the EBS-Ogna variant. One very large (Norwegian) family was reported to manifest this EBS subtype, characterised by haemorrhagic blistering and generalised skin fragility, sometimes mistaken as bruising [15].
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
Using Intravenous Immunoglobulins in Autoimmune Bullous Diseases
 Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
Living with Epidermolysis Bullosa: Reviewing the Impact on Individuals’ Quality of Life
 Dermatitis Herpetiformis
Dermatitis Herpetiformis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


