Major factors
Facial pain/pressure
Facial congestion/fullness
Nasal obstruction/blockage
Nasal discharge/purulence/discolored postnasal drainage
Hyposmia/anosmia
Purulence in nasal cavity on examination
Fever (acute rhinosinusitis only)
Minor factors
Headache
Fever (nonacute rhinosinusitis)
Halitosis
Fatigue
Dental pain
Cough
Ear pain/pressure/fullness
Acute Rhinosinusitis
Most episodes of sinonasal symptoms are caused by viral infections, which are a precipitator of ARS. A classic history for ARS is a week of nasal congestion and rhinorrhea that improves slightly and then returns with worsening symptoms several days later. This transition from viral rhinosinusitis to bacterial rhinosinusitis is variable and has been estimated to occur in only 0.5–2 % of cases [25]. Common bacterial pathogens associated with ARS have been identified as Streptococcus pneumonia, Haemophilus influenzae, Moraxella catarrhalis, and Staphylococcus aureus [26] (see Fig. 5.1). Most patients with ARS are successfully treated with broad-spectrum antibiotics and nasal care, although Rosenfeld et al. observed spontaneous resolution in 62–69 % of patients [24]. By definition, ARS manifests fewer than 4 weeks of clinical symptoms [9], after which the disease process is classified as subacute rhinosinusitis.
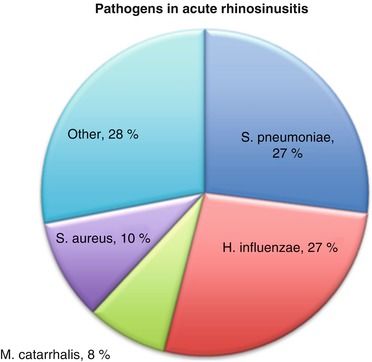
Fig. 5.1
Pathogens in acute rhinosinusitis (Reprinted from Payne and Benninger [26] by permission of Oxford University Press)
Chronic Rhinosinusitis
Symptoms that persist beyond 12 weeks despite maximum medical therapy are defined as CRS [20, 24]. Culture data from patients meeting the diagnostic criteria for CRS demonstrate a preponderance of Staphylococcus aureus, gram-negative species, as well as anaerobes [27]. All cases of rhinosinusitis (acute, subacute, and chronic) were initially believed to be infectious in nature, which has led to the prominent use of antibiotics and surgical drainage procedures as therapeutic modalities [2, 3]. However, it is likely that CRS is a chronic mucosal inflammatory process complicated by chronic infection.
The diagnosis of CRS requires careful consideration and integration of the details of the history, nasal endoscopy, and imaging data. The clinical history should elucidate the specific symptoms described by the patient and identify a pattern of symptoms, as well as precipitating or avoidable factors. Additionally, the previous response to medical therapy, coexisting conditions, a detailed environmental history, and home and occupational exposures constitute important pieces of information [9]. The clinical history can also be very powerful. Benninger showed that the history may be more indicative of CRS than nasal endoscopy and recommended giving it more consideration if the diagnosis is unclear or if limited anterior rhinoscopy is all that is available to the treating physician [28].
Nasal endoscopy is an important part of the physical exam and is typically performed with a rigid nasal endoscope. For patients meeting clinical symptom criteria for CRS, nasal endoscopy has been shown to significantly improve diagnostic accuracy [29]. Typical endoscopic findings of CRS are pus or polyps [24]. One study reported that pus was only present in patients with CRS and never present in patients with negative radiographic computed tomography (CT) findings [30]. Rosbe and Jones found that 91 % of patients with positive endoscopic findings of CRS had CT scans consistent with CRS [23]. Dykewicz and Hamilos found nasal endoscopy was sufficient to establish the diagnosis of CRS but not sufficient to establish the extent of the disease unless the patient had undergone a previous sinus surgery [9]. In a study by Stankiewicz and Chow, positive endoscopic findings did not often correlate with positive CT findings, but the absence of endoscopic findings showed 78 % correlation with negative CT findings [31]. Despite some discrepancies, nasal endoscopy is an important part of the clinical exam and gives the practitioner the opportunity to culture or biopsy suspicious findings [32]. It can confirm CRS but by itself cannot rule out a diagnosis of CRS.
The imaging modality of choice for the diagnosis of CRS is a non-contrast CT scan. The most commonly used sinonasal grading metric for the evaluation of sinonasal CT scans with a high degree of interobserver reliability is the Lund–Mackay score [33]. Radiographic evidence of CRS is important to the practitioner because it identifies the diseased sinuses and can help identify other contributing factors. For example, CT scans have been shown to be more accurate in diagnosing dental disease as the cause of maxillary sinusitis than a panorex, and more than 80 % of CT scans with a maxillary sinus fluid level greater than two thirds the height of the sinus with mucosal thickening have been shown to harbor dental pathology [34]. CT scans can also reveal other precipitating processes like benign sinonasal masses. They are not, however, recommended for the diagnosis of ARS unless a patient manifests with extra-sinus involvement [24]. Some discrepancies between the radiographic findings and patients’ reported symptoms have been reported. Specifically, CT scan findings do not always correlate with the severity of nasal symptoms [35, 36], and Zheng et al. found that radiologic severity assessed by the Lund–Mackay score is weakly correlated by CRS severity as measured by patients’ symptoms [37]. Despite these discrepancies, CT findings are still useful in that they are objective [38] and provide an important road map for surgical planning. Still, it is necessary when considering a diagnosis of CRS to consider the composite of patient’s history, nasal endoscopic findings, and radiographic data to make an accurate diagnosis.
Differential Diagnosis
The differential diagnosis for CRS is broad. A patient’s age is an important stratifier because some disease processes with similar symptoms are more common in children than adults (i.e., adenoid hypertrophy) [1, 9]. Children are also more likely to have nasal foreign bodies that may mimic CRS [1, 9]. Sinonasal tumors [1] like inverted papilloma, juvenile angiofibroma, antrochoanal polyps [9] and odontogenic sinus disease [34], and nasal carcinoma can present with similar symptoms as CRS. Anatomic abnormalities that predispose to recurrent sinus infections like nasal septal deviation may coexist with CRS and can mimic the symptoms of CRS [9].
Embryologic remnants like Thornwaldt cysts can also present similarly to CRS [1]. Furthermore, there is considerable overlap between sinus headaches and migraine or midfacial pain syndrome that can present with nasal congestion and rhinorrhea [39, 40]. Finally, systemic diseases like granulomatosis with polyangiitis (Wegener’s), sarcoidosis, and Churg–Strauss disease can have prominent sinonasal manifestations. Thus, a comprehensive differential diagnosis with appropriate workup is paramount to accurately diagnose CRS.
Pathophysiology of Chronic Rhinosinusitis
The etiology of CRS is the subject of much current research. Because different forms of CRS have different pathophysiology, no single process or pathogen has been identified as the trigger for CRS [4]. Still, some of the immunologic factors involved in CRS are well understood. Histologic findings have furthered the immunologic understanding of disease processes. Other contributing factors like superantigens and nitric oxide have been suggested. More recent data demonstrates that pulmonary physiology in asthma has substantial similarities to sinonasal physiology in CRS [41]. All processes result in inflammatory changes that lead to decreased sinonasal mucociliary clearance and perpetuate the disease process. Instead of a coordinated beating action by millions of cilia that line the surface of the sinus epithelium and move particles toward the natural ostia, swelling and inflammation compromise this process and result in a decreased ciliary beat frequency. This in turn leads to stagnation of sinus secretions, lack of oxygen in the sinus, and further reduction of mucociliary clearance and nitric oxide production [27].
Because all forms of CRS manifest varying degrees of inflammation, an understanding of the immunologic processes driving them is critical to treating the disease. Allergy is a well-known risk factor for CRS. Consequently, patients with CRS and allergic rhinitis have been shown to have elevated levels of interleukin-5 (IL-5) [42]. Elevated levels of IL-5 have been discovered in the serum and secretions of patients with allergic rhinitis as well as that of non-allergic nasal polyps [43]. Aurora et al. showed that peripheral blood leukocytes isolated from patients with CRS responded to control lavage samples to produce IL-5. The same lavage samples when applied to leukocytes from healthy controls showed no production of IL-5 [44]. Interleukin-1 (IL-1), a cytokine responsible for increasing the presence of adhesion molecules that assist in neutrophil recruitment, has also been shown to be elevated in CRS [45–48]. CRS has also been associated with increased bone marrow eosinopoesis [42] leading to increased serum IgE, particularly in allergic forms of the disease [43]. Interleukin-8 (IL-8), another powerful cytokine whose expression has been elevated in CRS that is generated by neutrophils, has been found elevated in non-allergic forms of CRS [49]. It functions as a chemoattractant for other inflammatory mediators [45–48], and levels fivefold higher than healthy controls have been documented in nasal polyps and turbinates of patients with CRS [50]. The complex inflammatory milieu of CRS has received much attention and may hold the key to fully understanding the disease process.
Histologic findings of CRS have demonstrated that the epithelium and basement membrane of patients with CRS are thicker than that of healthy epithelium [51, 52], and one study reported that this finding was more pronounced in children [53]. Saitoh et al. reported that in patients with CRS and nasal polyps, the thickness of the basement membrane and epithelium correlated with the number of infiltrated eosinophils [52]. Other studies have also demonstrated increased tissue eosinophils in CRS [51, 54, 55]. These histologic sinonasal findings are similar to those found in pulmonary tissue of asthmatics [56–58], and they have been found to be unaffected histologically based solely on a patient’s asthma status [52]. Additionally, tissue eosinophils have been found to be markedly increased in the sinonasal mucosa of asthmatics [59]. In asthmatics, eosinophilic infiltration was identified inside the subepithelial layer, and the amount of infiltration was related to the amount of epithelial damage and basement membrane thickness [59]. The amount of neutrophils was also increased in the subepithelial layer of the CRS sinonasal mucosa of these patients [59]. The histologic findings of the CRS mucosa suggest a link between upper and lower airway pathophysiology.
There are many similarities between asthma and CRS. The cytokine profile of CRS is similar to that of the lungs in asthma [57]. In fact, the inflammatory changes with epithelial shedding and thickening of the basement membrane identified in CRS are similar to the findings in asthmatic pulmonary tissue [51]. The discovery of neutrophils in CRS mirrors their discovery in asthma [4]. There are also clinical correlations between the lungs and sinuses indicating a link between the two. It has long been known that upper respiratory tract infections can precipitate asthma attacks [60]. Some have suggested that nasal obstruction, stasis of nasal secretions, and infections of the sinonasal mucosa may be a trigger for lower airway pathology in susceptible individuals [61].
Grünberg et al. demonstrated that after nebulization of a rhinovirus-16 suspension, asthma patients develop rhinitis symptoms with worsening of the asthma state [62]. Additionally, most patients with CRS who did not carry a diagnosis of asthma showed bronchial hyperreactivity when given a methacholine challenge [51]. Furthermore, the frequency of asthma in nasal polyp patients ranges from 17 to 31 % [63, 64], and the incidence of nasal polyps in asthmatics over age 40 years is 10–15 % [65]. Surgical treatment of CRS has been shown to have beneficial effects on asthma. Senior et al. demonstrated that after a mean of 6.5 years of follow-up, 90 % of asthmatics who underwent endoscopic sinus surgery (ESS) reported improvement in asthma, a lower number of asthma attacks, and less asthma medication usage [66]. Asthmatic children who underwent ESS reported fewer asthma-related hospitalizations and school days missed [67]. Finally, patients with CRS, nasal polyps, and asthma who underwent ESS had improvement in pulmonary function tests and required fewer systemic steroids for asthma control [65]. These studies have led to the conclusion that ESS in asthmatics is most often successful for sinonasal symptoms and may improve bronchial asthma symptoms as well as decrease medication use [68].
Nitric oxide (NO) has also been under investigation as a contributor to CRS [60]. NO is produced by the sinuses and functions as an innate immune mediator capable of killing bacteria, fungi, and viruses [27]. In cases of severe nasal obstruction, NO levels have been shown to be low, whereas inflammatory conditions are associated with a higher level of NO [69]. The clinical utility of NO has yet to be defined, but it has been proposed that it may be useful in assessing the response to treatment in CRS by gauging ostiomeatal complex (OMC) patency [70].
Biofilms have also been implicated as a cause of CRS [71], particularly refractory CRS. Biofilms are an organized community of bacteria adherent to a mucosal surface or foreign body and are situated in an extensive extracellular polymeric substance that is composed primarily of polysaccharides forming a glycocalyx [72]. This glycocalyx is a mixture of bacterial colonies of various phenotypes and serves as protection for its bacterial inhabitants while also modulating the microenvironment of the colonies through its numerous water channels via a process of interbacterial signaling called quorum sensing [72]. When in the form of a biofilm, infectious bacteria are difficult to detect and culture using conventional methods and are largely resistant to current antimicrobial therapy [73]. Antibiotics freely penetrate the bacterial biofilm, but the resistance is probably related to the slow growth conditions within the biofilm and sharing of multiple resistance genes within the members of the biofilm community [73]. Unfortunately, there is currently no simple, noninvasive clinical test for detecting biofilms; their identification relies on electron or confocal scanning laser microscopy or indirectly by identification of a DNA signature for presence of biofilm-forming genes [73]. In fact, in 2004, they were first discovered in sinonasal mucosa by using a scanning electron microscope to look at the mucosa from clinical nonresponders with CRS [71]. Since then, biofilms have been implicated in recurrent adenotonsillar infections, otitis media, and cholesteatoma [74, 75] and are thought by some to be present and likely contributors to all forms of CRS [76]. They have been identified in at least one third of CRS patients [73] and act as a source of bacteria which can be sources of antigens, superantigens, toxins, and other proinflammatory factors [76]. The most common bacteria identified in sinonasal biofilms is Staphylococcus aureus, but Pseudomonas aeruginosa and Haemophilus influenzae have also been identified [77–80]. Unfortunately, the presence of biofilms prognosticates a poor outcome after surgery [81, 82]. The presence of a biofilm does not determine the clinical course of the disease, but the persistence of the pathogenic organism via biofilm-forming capacity can impact the disease course [73]. Treatment for biofilms involves mechanical pressure irrigation [83] and different topical therapies [73]. Surfactant solutions have been tried with some success [84], and some data suggests that sinus rinses that contain baby shampoo have some effect [85]. Mupirocin irrigations have also been found to be effective for staphylococcal-cultured positive biofilms [86]. Treatment often requires prolonged courses of topical modalities after shorter courses of other medical and surgical therapies.
Other contributors to CRS have been proposed. Superantigens have been described in the literature, and enterotoxins from Staphylococcus aureus, a well-known pathogen in CRS, have been hypothesized to cause some forms of CRS [87]. Bachert et al. demonstrated that the presence of Staphylococcus aureus enterotoxin was associated with the upregulation of the production of polyclonal IgE antibodies [88]. Some suggest that hypoxia may be the etiology of CRS [89, 90]. Recently, it was hypothesized that fungi were responsible for CRS. It was thought that toxic mediators secreted by eosinophils that play an essential role in the elimination of sinonasal fungal infections also have an unwanted side effect of causing local tissue destruction and CRS-related symptoms [91, 92]. Later, a placebo-controlled study did not show any relevant effectiveness of antifungal treatment in the alleviation of CRS symptoms or on relevant mediators [93–96].
Risk Factors for Chronic Rhinosinusitis
Many factors contribute to the development of CRS. There is a tendency for CRS to run in families [97]. Cohen et al. reported that the severity of the disease process is proportional to the penetrance of an underlying genetic component [97]. Aspirin intolerance [98] and diseases that cause decreased mucociliary clearance [99] predispose to the development of CRS. Some forms of CRS are precipitated by anatomic deformities, specifically nasal septal deviation, concha bullosa, paradoxical middle turbinates, or Haller cells [9]. However, these anatomic variants are also seen in patients without CRS [100–103]. Smoking [104] and gastroesophageal reflux (GER) [1] have been associated with CRS. Smoke has been shown to inhibit the mucociliary clearance and epithelial regeneration of sinonasal mucosa [105], and GER has been associated with CRS, but direct causality has not been demonstrated [106].
Atopy has long been associated with CRS [104]. Although it is not causal in its relationship, there is a correlation between allergy and CRS. Newman et al. demonstrated that an increased serum total IgE level was associated with severe CRS [107]. Rachelefsky et al. reported that 53 % of people with allergic rhinitis had sinusitis [108], and other reports estimate that 25–58 % of people with sinusitis had allergic rhinitis [109, 110]. Allergic rhinitis without CRS has been shown to negatively impact a patient’s quality of life compared to the general level of the US population [111]. Atopy with CRS is also a predictive factor for decreased quality of life and poorer surgical outcome for CRS patients [1]. CT scans in atopic CRS patients are also more likely to show increased inflammatory changes [107, 112, 113]. Steinke and Borish reported that more than 50 % of people with perennial allergic rhinitis have been shown to have abnormal CT scans [108, 114]. Berrettini et al. compared CT scans from adults with perennial allergic rhinitis with controls and found that 67.5 % of the atopic group had evidence of CRS compared with 33.4 % of controls [115]. CRS patients who required ESS were also more likely to be atopic. Tan et al. reported that 82 % of patients with CRS requiring ESS had positive skin tests which was similar to patients with allergic rhinitis (72 %) [116]. Emanuel and Shah showed that 84 % of patients who failed maximal medical therapy and required ESS tested positive for allergy [117]. Interestingly, more than 60 % of them had house dust mite allergy. It has also been reported that the most prevalent positive skin test in CRS and allergic rhinitis was to dust mites and that sensitivity to multiple allergens and perennial allergies put patients at a higher risk for chronic hyperplastic eosinophilic sinusitis [116]. Therefore, some have suggested that perennial allergy may be one of the underlying inflammatory factors for CRS [118]. Although some data suggests that atopic status is weakly associated with the severity of CRS [119–121], there is general consensus that management of allergy in CRS is important for proper disease control [1].
The mechanism by which allergy contributes to CRS is complex. Bacterial ARS can be due to allergic rhinitis, especially as mucosal inflammation causes occlusion of the sinus ostia with stagnation of secretions within the sinus cavities [109]. Moreover, the mucosal changes due to allergic rhinitis can alter mucociliary clearance, which can further predispose to CRS [1]. But this mechanism appears too simplistic to describe the relationship between CRS and allergy. Nasal allergen challenges in allergic patients have been shown to yield increased eosinophils and histamine in nasal and maxillary sinus specimens [122]. Furthermore, nasal allergen challenges in sensitive patients have demonstrated radiographic opacification in the maxillary sinus [123]. In 2004, Steinke and Borish described a potential mechanism between atopy and CRS that explains many of the observed relationships [124]. Environmental peptides are loaded onto dendritic cells associated with sinonasal cavities and migrate to nasal-associated lymphatic tissue. T-helper cells migrate from the nasal airway to the nasal lymphatic tissue and bone marrow. Some of the cells newly activated in the nasal airway may include locally produced eosinophil precursors. Once delivered to the bone marrow, these Th2-like cells stimulate the production of inflammatory cells, including basophils, eosinophils, and mast cell precursors. These inflammatory cells infiltrate susceptible tissues like sinuses and lungs. A process of selective recruitment takes place whereby tissues are induced to express appropriate adhesion molecules for inflammatory mediators. This process only occurs in the presence of preexisting disease. Subjects without preexisting sinusitis (or non-asthmatics) do not have adhesion molecules in their airways so their exacerbations of rhinitis do not spread to the sinuses (or lungs). Therefore, allergen-induced rhinitis or non-allergic rhinitis can cause increased systemic inflammation that may contribute to exacerbations of asthma frequently seen in individuals with these underlying conditions.
Leukotrienes have also been implicated in the pathology of allergy. Cysteinyl leukotrienes (cysLTs) have proinflammatory capabilities. Specifically, they induce chemotaxis, increase eosinophilic inflammation of the airways, increase smooth muscle hyperreactivity as well as vascular permeability, and increase mucous secretion, thereby reducing mucociliary clearance [125]. They have been found in elevated levels in the nasal polyps of patients with chronic hyperplastic eosinophilic sinusitis compared to tissue from patients with chronic inflammatory sinusitis and healthy sinus tissue [125].
Asthma is a well-known risk factor for CRS [41]. The incidence of asthma in CRS patients is 23 % compared with 5 % in the general population [126], and CRS has been estimated to coexist with asthma in 30–50 % of patients [127, 128]. Ponikau et al. demonstrated that up to 91 % of patients with CRS had asthma or bronchial hyperreactivity [51]. Many patients with asthma have also been shown to have radiographic findings suggestive of CRS. In adult asthmatics, 74–90 % had CT evidence of some degree of mucosal hyperplasia [107, 129, 130], and in patients with CRS and asthma, a higher Lund–Mackay score was associated with more severe asthma [38, 113, 131, 132]. Two forms of asthma have been described: a Th2 eosinophilic form and a Th2 non-eosinophilic form [59]. Similarly, asthma has been associated with an increase in eosinophils [59] that often correlates with the severity of asthma [133, 134] and a neutrophilic pulmonary infiltrate (rather than eosinophilic) [59]. Similar histologic observations have been made with CRS. Asthma with CRS is also associated with severe nasal polyposis [134].
A dual diagnosis of asthma and CRS has many clinical implications. While asthma alone was not shown to negatively impact a patient’s quality of life, asthma with nasal polyps has been demonstrated to severely affect one’s quality of life [12, 135]. In 2008, Staikuniene et al. reported that patients with CRS, asthma, and nasal polyps were more likely to be of older age, have a greater duration of nasal symptoms, undergone more previous sinus surgeries, present with worse CT scans, have a higher blood leukocyte and eosinophil count, have higher total IgE level, have increased bronchial obstruction, and have a higher incidence of allergic rhinitis [118]. Many studies also suggest that treatment of CRS in asthmatics improves asthma severity [38, 136–138]. In a study of 30 patients who had undergone FESS, 90 % reported asthma improvement, 74.1 % had fewer asthma attacks, 46 % described less inhaler usage, and 65 % reported decreased steroid requirement [66]. While the physiologic mechanism linking asthma to CRS is still debated, some potential mechanisms have been suggested [139]. Some have suggested that inflamed sinus secretions that are aspirated into the lower airways could precipitate an asthma attack. Others have proposed that enhanced vagal stimulation in the infected sinus could lead to bronchospasm. Finally, inflamed sinuses may produce cytokines that act as bronchoconstrictive mediators in the lower airways. As the mechanism is more completely defined, the treatment modalities for both CRS and asthma will hopefully improve. For now, it is clear that optimal treatment of CRS requires addressing a patient’s asthma as well.
A common manifestation of immunodeficient patients is CRS. Some estimates report that 8–20 % of cases of persistent or recurrent ARS are due to immunodeficiency [1]. Similarly, deficient antibody production in response to vaccination or hypogammaglobulinemia was found in 12 % of adults with CRS with nasal polyps [140]. Therefore, chronic recalcitrant sinusitis should raise suspicion about the possibility of immunodeficiency [27]. Typically, these patients present with sinonasal symptoms that are responsive to antibiotic therapy but recur after their withdrawal [141]. This presentation is also common to other forms of CRS, so low index of suspicion is required. The most common primary immunodeficiency in adults is common variable immunodeficiency. It occurs in up to 10 % of patients with refractory CRS. General immunoglobulin deficiencies are seen in as many as 22 % of refractory CRS patients [142, 143]. Table 5.2 demonstrates appropriate lab tests to begin a workup for immunodeficiency. A multidisciplinary approach should be undertaken for these patients.
Table 5.2
Immunodeficiency evaluation
Complete blood count with differential |
Quantitative immunoglobulins: IgA, IgE, IgG, IgM |
Immunoglobulin subclasses: secretory IgA, IgG1, IgG2, IgG3, IgG4 |
T-cell subpopulations: CD4, CD8 |
Pneumococcal antibody titers: before and 6 weeks after pneumococcal vaccination |
Classifications of Chronic Rhinosinusitis
The classification systems of CRS have mirrored our understanding of the disease processes that cause sinonasal inflammation. Much effort has been spent developing and refining different schema with a goal to improve patient outcomes. This is due in part to the difficulty associated with managing refractory CRS. Initial classification systems stratified CRS based on the presence or absence of nasal polyps [56, 144–147], and significant differences were discovered. CRS without nasal polyps demonstrated elevated levels of Th1 and Th2 mediators [145, 148, 149], with higher levels of neutrophils, IL-1, and IL-8 [45–48]. Furthermore, no difference was observed in epithelial IgE levels between CRS patients without nasal polyps and healthy controls [150]. On the other hand, CRS with nasal polyps demonstrates an immune profile with a Th2 skew with elevated levels of mast cells [151], IL-4 and IL-13 [152], and typically a preponderance of eosinophils [145, 148, 149, 153] with fewer observed neutrophils [153]. The prevalence of CRS with nasal polyps is estimated to be approximately 30 % in patients with CRS [38]. Approximately 80–90 % of all cases of nasal polyposis are characterized by eosinophilic infiltrates [154], particularly in Western cultures [155]. Curiously, Asian nasal polyps tend to be more neutrophilic [156] although they are macroscopically indistinguishable from their Caucasian counterparts [4]. Even in non-Asians, some nasal polyps do not contain elevated levels of eosinophils [157, 158]. Patients with CRS with nasal polyposis are also more likely to be associated with asthma [41, 159, 160]. Up to 7 % of asthmatics have nasal polyps [68], and CRS patients with nasal polyps are more likely to have aspirin sensitivity or asthma than the general population [1]. Although this classification provided some clinical value, it was still viewed by many as overly simplistic and incomplete.
Additional subclassification schemas have been introduced. Steinke and Borish [124] divided CRS into four subsets: CRS due to immunodeficiency, inflammatory CRS without prominent hyperplasia of immune cells, chronic hyperplastic eosinophilic sinusitis (CHES), and allergic fungal sinusitis (AFS). Two more categories were added later—aspirin-exacerbated respiratory disorder (AERD) and non-eosinophilic CRS [27]. CRS associated with immunodeficiency, AFS, and AERD are discussed later in this chapter. CHES is similar to most forms of CRS with nasal polyposis. CHES is often characterized by nasal polyposis [57, 161–165], activated eosinophils, fibroblasts, mast cells, and goblet cells [19, 161, 166]. The cytokine profile is also similar in that IL-3, IL-4, IL-5, IL-13, eotaxin, and GM-CSF levels are elevated [125].
Non-eosinophilic Sinusitis
Non-eosinophilic sinusitis (NES) is a more recently characterized category of CRS [167]. These patients do demonstrate levels of eosinophilia that are higher than controls, but lower than CHES [167]. The disease is due to chronic or recurrent occlusion of sinus ostia by viral rhinitis, allergic rhinitis, or anatomic predisposition [27, 145] and often predisposes to the formation of biofilms [158, 167]. Patients with NES present with recurrent and protracted sinonasal bacterial infections that cause barotrauma to the sinus cavity that in turn damages the respiratory epithelium resulting in ciliary dysfunction and mucous gland and goblet cell hyperplasia [27]. NES with nasal polyps shows similar histologic findings with a large mononuclear cell infiltrate, fibrosis, and mast cells [167, 168]. Mast cell concentration is increased in sinonasal connective tissue [167], and there is an interplay between fibroblasts and mast cells [169, 170] that results in more fibroblast recruitment and collagen deposition [27]. B-cell and plasma cell expression appears to be upregulated [158, 171]. The basement membrane of NES sinonasal mucosa is histologically thinner than that in eosinophilic nasal polyps [172], and it takes place in the absence of allergic disease [152]. Its pathophysiology is less due to Th1- or Th2-mediated processes; NES is thought to be more related to an innate immune response than atopy [152]. This is supported by the findings that lymphocytes expressing CCR5 (a Th1 marker) and CCR3 (a Th2 marker) were less frequently observed in NES than in eosinophilic sinus disease [172].
The cytokine profile of NES with nasal polyps is also markedly different than that of eosinophilic nasal polyps. In NES, there is no upregulation for genes associated with IL-4 and IL-13, two cytokines that are associated with eosinophilic allergic inflammation [152]. CXCL1, a cytokine with neutrophil chemoattractant activity, is upregulated in NES as are IL-6, IL-8, monocyte chemoattractant protein 1, and hypoxia-inducible factor-1α (HIF-1α) [152], an inducible transcription factor expressed in hypoxic conditions that is involved in the activation of glycolytic and inflammatory pathways [173]. NES has also been shown to be associated with decreased expression of cysLT2R protein [174], but some studies report that cysLT levels in NES sinonasal mucosa are similar to controls [125]. Tenascin-C is a cytokine that is expressed in transient acute tissue injury [175]. Its levels are elevated in NES with nasal polyps, and in the presence of chronic disease, it implies persistent acute injury; this may be responsible for the fibrotic characteristic of polyps associated with NES [175]. As the ostial obstruction is surgically corrected, the hypoxic conditions and resultant inflammation resolve, resulting in less barotrauma of the sinuses. Although much still remains to be learned about NES, it often appears to be a potentially surgically treatable disease.
In 2013, the senior author proposed a subclassification system that stratified patients based on two clinical variables, the presence of allergy and asthma [176]. This resulted from a prospective case–control study in which histopathologic findings and immunohistochemistry results of nasal polyps obtained from 84 patients were compared with nasal endoscopic findings and sinonasal CT scans. The result was the creation of seven subclasses of CRS (see Table 5.3): aspirin-exacerbated respiratory disease (AERD aka aspirin triad), asthmatic sinusitis with allergy (AScA), asthmatic sinusitis without allergy (ASsA), non-asthmatic sinusitis with allergy (NAScA), non-asthmatic sinusitis without allergy (NASsA), allergic fungal sinusitis (AFS), and cystic fibrosis (CF). Although this classification system may be refined in subsequent studies, it is useful because it allows a patient to be characterized based on their clinical picture (phenotype) that can then be correlated to the immunohistochemical profile (endotype).
Aspirin-exacerbated respiratory disease |
Asthmatic sinusitis with allergy |
Asthmatic sinusitis without allergy |
Non-asthmatic sinusitis with allergy |
Non-asthmatic sinusitis without allergy |
Allergic fungal sinusitis |
Cystic fibrosis |
NASsA is characterized by endoscopic purulence without high CT scores. These patients often have a structural abnormality predisposing to persistent bacterial infections. Biofilms are not uncommon and eosinophils are sparse. Treatment involves oral antibiotics, steroids, and ESS for those who fail medical therapy. NASsA is similar to “inflammatory CRS without prominent hyperplasia of immune cells.”
NAScA is mediated by helper T cells. It is driven by a combination of infectious and inflammatory processes and demonstrates a higher amount of eosinophils and mast cells than controls. The cause is thought to be due to an acute allergy exacerbation that led to mucosal swelling and often resulted in a cyclical sinusitis pattern. Treatment involved pharmacotherapy addressed at the allergy component of CRS.
AScA has been described in the literature as a “unified airway,” in which sinusitis, allergic rhinitis, and asthma are present, but not aspirin sensitivity. These patients commonly have a pediatric history of allergy or asthma and often present with extensive nasal polyposis with little purulence on endoscopy. The pathophysiology of AScA is driven by a Th2 process, and eosinophils levels are very high in these nasal polyps. The connection between upper and lower airway has been proposed to involve the circulation of eosinophils that are activated in the sinus mucosa and then transported to the lungs via the circulatory system where they bind to adhesion molecules in pulmonary epithelial tissue and create a local inflammatory response. Treatment typically involves surgical debulking of the polyps with medical therapy in the form of topical budesonide and immunotherapy. AScA resembles “chronic hyperplastic eosinophilic sinusitis.”
ASsA is thought to be a precursor to AERD. Patients typically do not have a history of pediatric atopy or asthma, and asthma in ASsA patients typically develops when they are adults. These patients should be counseled to avoid the use of NSAIDs and should undergo a workup for AERD. Management is similar to that of AERD.
Allergic Fungal Sinusitis
The first reported patient with AFS was thought to be suffering from allergic bronchopulmonary aspergillosis (ABPA) and had symptoms of nasal obstruction; hard, blood-tinged nasal casts; and nasal polyps. Sinonasal cultures demonstrated Aspergillus fumigatus [177]. In 1981, the sinus contents of these patients were compared to the bronchopulmonary mucus plugs characteristic of ABPA, resulting in the development of the term “allergic aspergillosis” to describe AFS [178]. In 1983, the allergic mucin of AFS was histologically compared to the pulmonary mucoid impactions and found to be identical [179]. Fungal hyphae were identified that were similar to those of Aspergillus; therefore, the term “allergic Aspergillus sinusitis” was coined. With the identification of other fungal species, the name was changed to “allergic fungal sinusitis,” by which the disease process is currently known [180].
AFS typically affects adults with mean ages between 20 and 35 years [181], although it has been documented in children [99]. Those affected are immunocompetent [181], and men and women are equally affected [181]. In South Australia, it was estimated that 4–10 % of patients undergoing ESS have evidence of AFS [182]. The true prevalence of the disease is unknown because diagnosis requires examination of the surgical specimen and certain geographic locations are more affected by the disease [181]. In the United States, the humid river basins and coastal regions of the southeastern states are the most common locations to see AFS [150].
The signs and symptoms of AFS are also unique to the disease process. Patients with AFS present with a long-standing history of nasal obstruction, hyposmia or anosmia, and blowing out nasal casts [181]. Often the nasal obstruction is unilateral [27]. It is characterized by thick eosinophilic mucin, nasal polyps, and often painless proptosis and diplopia, with or without epiphora [181], especially in children [99]. In 1994, Bent and Kuhn defined the diagnostic criteria for AFS that are still used today [183] (see Table 5.4). Patients with AFS demonstrate a type 1 hypersensitivity, with positive sinonasal fungal stains/cultures, characteristic radiographic findings on CT, eosinophilic mucin without fungal tissue invasion, and nasal polyposis. The physiology of type 1 hypersensitivity depends on permanent sensitization of mast cells in the target organ where a high-affinity receptor of IgE is located. Specific allergens cause the receptors to cross-link, which results in the release of preformed and newly formed mediators that cause local inflammation and recruitment of leukocytes, including basophils and eosinophils, leading to the clinical symptoms and signs of the disease [181]. This is an exaggerated or inappropriate immune-mediated reaction to an antigen at a dose tolerated by normal subjects [184]. Diagnosis of type 1 hypersensitivity requires the detection of allergen-specific IgE in peripheral blood or a positive skin prick test to that allergen [185]. In AFS, up to 100 % of patients have fungal allergy [183, 186, 187], but not necessarily to the same species identified on culture [181].
Table 5.4
Bent and Kuhn criteria for allergic fungal sinusitis
Type 1 hypersensitivity |
Positive fungal stain/culture |
Characteristic radiographic findings |
Presence of eosinophilic mucin without fungal invasion |
Nasal polyposis |
The diagnosis of AFS requires the identification of fungi on culture or stains. Common fungi implicated with AFS include Aspergillus, Alternaria, Bipolaris, Cladosporium, Curvularia, Drechslera, and Helminthosporium [181]. Yet the mere presence of fungi does not imply an allergic response. The average person inhales millions of fungal spores daily. Furthermore, fungal allergy is not synonymous with AFS. In fact, a significant proportion of CRS patients have fungal allergy but do not have AFS [181]. Fungal allergy is estimated to affect 3–10 % of adults and children worldwide [188]. It may coexist with AFS and may exacerbate an underlying inflammatory disorder rather than be a major contributor to its pathogenesis [181].
Eosinophilic mucin is a peculiar finding in AFS. Macroscopically, it is thick, viscous to almost solid, and colored. Often it is described as axle grease, peanut butter, or cottage cheese-like consistency [189]. It contains eosinophil breakdown products known as Charcot–Leyden crystals, other leukocytes, respiratory epithelial cells, and debris [190]. In 4–10 % of CRS patients with eosinophilic mucin in certain high-prevalence regions, fungal allergy is also present [182]. At this time, “it remains unclear whether CRS patients with eosinophilic mucin but no fungal elements or fungal allergy represent a different clinical and pathological entity from AFS patients” [181]. Eosinophilic mucin has an important role in the prognosis of CRS and signifies a distinct form of CRS associated with intense mucosal inflammatory response, a worse clinical course, and greater prevalence of lower respiratory tract disease [181].
The radiographic findings associated with sinonasal CT scans of AFS patients are unique. AFS has been shown to cause sinonasal expansion into adjacent structures and bony remodeling [191] but is also characterized by “double densities” on CT scans [192] (see Fig. 5.2). These CT scans demonstrate heterogeneous signal intensity within the sinuses caused by an increase in heavy metals including iron, manganese, and calcium that are associated with fungi [181].
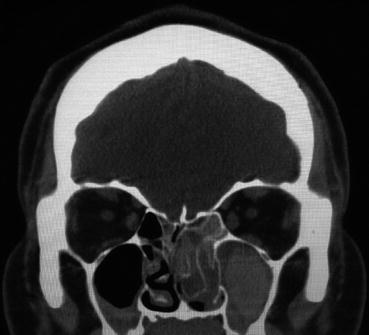
Fig. 5.2
Coronal non-contrast CT scan showing the “double densities” characteristic of AFS
Nasal polyps of AFS are thought to be due to fungi, enterotoxins, eosinophils, and IgE [88, 172, 193–195]. There are conflicting reports about the eosinophilia of the polyps themselves [88, 172].
The Bent and Kuhn criteria for AFS are still the gold standard in the diagnosis of AFS. More recently, variations in diagnostic criteria have been suggested. A recent international panel suggested that a CRS patient with histological confirmation of eosinophilic mucin and the presence of type 1 hypersensitivity meets the criteria for the diagnosis of AFS [144]. Others question the need to demonstrate fungal allergy and suggest that the macroscopic presence of eosinophilic mucin is sufficient [182, 196, 197]. Additionally, a subclassification was proposed for patients with eosinophilic mucin and fungal allergy who did not meet the remaining diagnostic criteria [181]. At present, the gold standard for the diagnosis of AFS remains the Bent and Kuhn criteria.
The pathophysiology of AFS is still incompletely understood. However, there is evidence to suggest a predilection toward a Th2 cytokine profile with increased eosinophils and fungal and nonfungal IgE [150, 181]. Specifically, there is an increase in total serum IgE, peripheral blood eosinophils, serum eosinophil cationic protein, erythrocyte sedimentation rate, and often C-reactive protein [198, 199]. It has been suggested that AFS is driven by a local production of IgE [88]. In allergic rhinitis patients, there is an increased local sinonasal mucosal production of allergen-specific IgE [200, 201], and one study found increased antigen-specific fungal and nonfungal IgE in the sinus mucosa of AFS patients compared to controls [202]. Histologically, the subepithelium of AFS sinonasal mucosa demonstrates more IgE staining than the epithelium [150].
Initial treatment of AFS focused on the infectious process without much success. Current recommendations direct treatment toward the control of the atopic mechanisms [189]. Despite the name, there is no role for topical or systemic antifungal therapy in the treatment of AFS [203]. Initial treatment is aimed at surgical removal of polyps and eosinophilic mucin characteristic of this process. Systemic steroids are an important adjunct to this process. After the initial ESS, maintenance of the disease is performed by topically administered steroids in suspension [204] in the forms of irrigations and sprays. The benefit of fungal immunotherapy is still unclear [205]. However, the benefit of immunotherapy with nonfungal inhalant antigens has not been evaluated in this population [206, 207]. The long-term successful treatment for AFS has not been clearly defined although the use of topical steroid irrigation has been described [181], and many patients require additional surgeries since the patients maintain the inflammatory allergic disease despite surgery [27].
Aspirin-Exacerbated Respiratory Disease
Also known as aspirin triad, Samter’s triad, or Widal’s syndrome, AERD is a complex disease in which a very small single dose of aspirin or other COX-1 inhibitor can cause severe symptoms [1]. Aspirin-like compounds have been used since the time of Hippocrates (approximately 400 B.C.), when the bark of the white willow was used as an antipyretic agent. It was also used during Roman times and again in the 1700s as a treatment for fever [208]. The chemical structure was described and subsequently modified in the 1800s to produce the stable compound acetylsalicylic acid, which is now called aspirin. Bayer released this onto the market in 1899 as an analgesic and antipyretic agent. Soon thereafter, it was recognized that severe asthma attacks could occur after ingestion of aspirin. In 1922, Widal et al. [209] described the clinical symptoms of aspirin sensitivity, asthma, and nasal polyps and subsequently performed the first aspirin desensitization. Later, Samter and Beers described and characterized the aggressive mucosal disease associated with aspirin sensitivity, leading to the name “Samter’s triad” [210].
AERD is an autosomal disorder [211, 212] with de novo development in adulthood [27] and a mean age of onset of 40–50 years [210]. Approximately 2–8 % of CRS patients with nasal polyps will also have AERD [120]. The classic triad of nasal polyps, asthma, and aspirin sensitivity characterizes AERD [210], although the disease develops gradually over time. Typically, patients will develop allergic rhinitis in their 30s, and 1–5 years later, they are diagnosed with asthma with aspirin sensitivity. Finally, within the next 5 years, they develop nasal polyps [213]. There are some variations to this process. For example, some patients may not develop asthma, while others may not manifest aspirin sensitivity during the initial progress of this disease. Therefore, a low index of clinical suspicion is required, especially in patients with recurrent polyps and intrinsic asthma with allergy that does not correlate to the severity of the atopic disease [69]. Additionally, there is a 30 % prevalence of aspirin sensitivity in people with asthma and nasal polyposis [214, 215], further necessitating a low index of suspicion.
AERD patients who ingest aspirin or NSAIDs classically develop a reproducible reaction within 20–120 min characterized by any of the following symptoms: facial flushing, perspiration, intense lethargy, rhinorrhea, nasal congestion, conjunctivitis, cough, bronchospasm, gastrointestinal symptoms, and even respiratory arrest and shock [27, 69, 213]. Once a patient develops aspirin sensitivity, they must avoid any NSAID, as aspirin sensitivity is often lifelong [216, 217]. Despite the “triad” of symptoms, patients with AERD seldom complain about sinus pressure or headaches [27]. Rather, anosmia is a more consistent complaint, and 65 % of aspirin-sensitive patients with CRS were reported as having olfactory impairment [27].
Asthma associated with AERD is a severe form that is often difficult to control and frequently associated with a progressive irreversible decrease in pulmonary function [218, 219]. Marquette et al. reported that 25 % of asthmatics requiring intubation for asthma had aspirin sensitivity. Ingestion of aspirin was not the cause of respiratory distress leading to intubation, suggesting that they may have an unstable disease despite appropriate avoidance measures [220]. Unfortunately, aspirin-induced asthma is not uncommon with estimates as high as one in ten asthmatics [60]. Aspirin-induced asthma is also present in up to 30 % of asthmatic patients with CRS and nasal polyps [27]. Non-AERD patients with aspirin-induced asthma also had a higher incidence of sinus disease, with one report suggesting an average of 5.5 episodes of sinusitis requiring antibiotics annually [217].
Nasal polyps in AERD are different from other types of polyps. They are more aggressive [27] and contain threefold higher concentration of eosinophils than other forms of polyps associated with CRS [167] and five times more eosinophils than asthmatic airways [221].
The pathophysiologic mechanism of AERD has been well defined (Fig. 5.3). Arachidonic acid is formed from the cell membrane. It can undergo conversion by cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) to prostaglandins and prostacyclins, or it can generate leukotrienes via the lipoxygenase pathway. Moorwood et al. described the physiology as follows [213]. COX-1 and COX-2 are constitutively expressed in the airway mucosa, and COX-2 is induced by proinflammatory signals. NSAIDs with COX-1 inhibitory activity all produce the aspirin reaction in sensitive individuals, but NSADs without COX-1 inhibition do not produce a reaction [222]. The reduction in COX-2 activity in aspirin-sensitive individuals, along with inhibition of COX-1 by aspirin, may together contribute to reduced prostaglandin-E2 (PGE2) production, resulting in clinical symptoms. There are some limitations with this model, but a decrease in PGE2 and COX-2 has been observed in AERD [163, 223] and PGE2 prevents activation of basophils, mast cells, and eosinophils. If a patient has a baseline deficiency of PGE2, they are susceptible to a massive inflammatory response [27]. Exogenously administered PGE2 has been shown to mitigate this response [224].
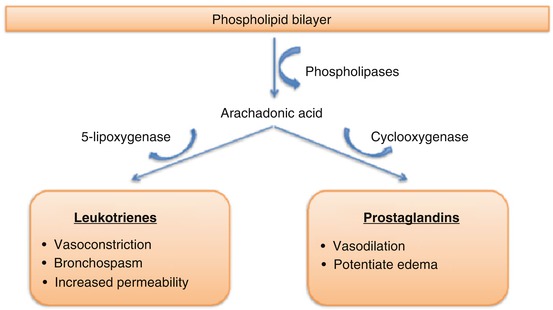
Fig. 5.3
Arachidonic acid pathway
Patients with AERD have an overproduction of, and over-responsiveness to, cysLTs [125, 163]. CysLTs are lipid mediators that not only stimulate potent contractive activity of bronchial smooth muscle but also exert proinflammatory actions both in the upper and lower airways [174]. They act on target organs through specific receptors [174], and they are overexpressed in AERD [225, 226]. This correlates with the increased number of sinus mucosa infiltrated eosinophils observed in AERD [174]. Sinonasal tissue in AERD also demonstrates upregulation of 5-lipoxygenase and leukotriene C4 synthase [163, 221, 227]. Finally, the percentage of inflammatory leukocytes expressing cysLT1R is dramatically increased in the mucosa of patients with aspirin-sensitive asthma compared to aspirin-tolerant controls, which may explain the increased responsiveness of target organs in these patients [174, 225].
The diagnosis of AERD requires a high index of suspicion. Not only should it be suspected in patients with recurrent nasal polyps and asthma, it should also be included in the differential diagnosis for asthmatics with severe asthma and chronic congestion with watery rhinorrhea, sudden severe asthma with intensive care unit admissions, and adult-onset non-allergic asthma [213]. Interestingly, AERD patients do not manifest atopy [213], and their total IgE concentration tends to be modest [27]. An appropriate screening test for AERD is a nasal challenge. It is easily administered but requires a 4-h observation period [213]. If results are negative, an oral challenge is indicated [69]. Therefore, many practitioners immediately employ the oral provocation test, which is the current gold standard [1]. In patients with a history of aspirin sensitivity, it is 85 % sensitive [213]. The mean time for reaction is approximately 85 min, with an average dose of 67.5 mg [213]. The oral provocation test should only be administered in physician offices with resuscitation capabilities [69].
Treatment for AERD can be very challenging for patients and practitioners. First-line therapy includes avoidance of all nonselective COX inhibitors [213] with the exception of patients undergoing desensitization who may tolerate different amounts of NSAID. First-line medical treatment includes leukotriene receptor agonists [27]. Although they do not block the reaction in aspirin-sensitive individuals, they can convert the reaction from a predominant bronchospastic event to symptoms involving the upper airways [213]. Lipoxygenase inhibitors have also been beneficial for the treatment of AERD. In aspirin-sensitive patients, zileuton has been shown to improve asthma, reduce the corticosteroid requirement, reduce nasal polyps, and restore anosmia [228]. ESS is used to remove sinonasal polyps, but without proper medical management after surgery, nasal polyps universally recur [229]. For patients who require additional therapy, desensitization may be indicated. Current indications for desensitization include asthmatic patients who can only be controlled with progressively increasing amounts of corticosteroids, patients with recurrent polyposis requiring repeat ESS, and patients who need aspirin or NSAIDs to treat rheumatic or thrombotic conditions [213]. Aspirin desensitization has been shown to decrease the production of cysLTs and the expression of cysLT receptors [225, 230]. Stevenson et al. demonstrated that desensitization improved asthma control, led to fewer required corticosteroid bursts, improved (and in some cases restored) the sense of smell, decreased the need for repeat polypectomies, and markedly decreased the occurrence of bacterial superinfections [231]. Other studies have shown that desensitization lessens both upper and lower airway symptoms of AERD but did not lead to complete remission [232]. Unfortunately, up to 30 % of patients cannot tolerate the side effects of daily aspirin therapy [1]. The most common side effect with high-dose aspirin therapy is gastrointestinal bleeding [69]. In those cases, oral doses as low as 100 mg daily may also be effective and decrease the risk of complications [233].
Although patients with AERD should avoid NSAIDs, there is some data to suggest that selective COX-2 inhibitors appear to be safe. Still, some suggest that the first dose be given with monitoring for 2 h in a facility where resuscitative capabilities are available [69]. Acetaminophen is not always tolerated in AERD patients, but Szczeklik et al. demonstrated that single doses of up to 500 mg appear to be safe in up to 94 % of AERD patients [234].
Cystic Fibrosis
CF is a unique form of CRS. In the past, it was mainly a pediatric disease, but with better pulmonary treatments, patients with CF are living well into adulthood. It is an autosomal recessive disease caused by a mutation in the CF transmembrane receptor chloride channel (CFTR) [235]. Those with complete absence of the CFTR gene have more acute and severe rhinosinusitis, while those with mutations resulting in a partially functional channel have a milder form of CRS [236]. CF afflicts approximately 1 in 3,500 live births [237], and the carrier frequency in the general population is approximately 1 in 25. Reports have suggested that up to 8 % of patients with CRS are heterozygotes for CF [238].
All patients with CF will eventually develop CRS [235] as the impaired mucociliary transport predisposes the sinuses to colonization of bacteria [235]. The most common form of CRS in CF is similar to NES that results from blockage of the ostia [235]. A vicious cycle ensues in which functional obstruction (due to poor mucociliary transport) results in inflammation and remodeling [235]. Secondary inspissated mucous and biofilms often develop [235], and mononuclear cells and activated B cells with secondary germinal center formation have also been identified [149, 167]. Increased levels of IL-5 are seen in CF [4] as are nasal polyps, which are often the presenting complaint of CF patients [239]. The polyps associated with CF are unique in that they rarely demonstrate eosinophilia; rather, neutrophilia predominates [4, 235]. Some have speculated that nasal polyps associated with CF represent a subtype of non-eosinophilic nasal polyps given the histologic similarity [235].
One unique characteristic of CRS associated with CF is the higher concentration of DNA that was identified in CF mucin [235], along with the elevated concentration of IL-8 and neutrophils [145]. Steinke et al. [235] explained that extracellular DNA is derived from granulocytes, secreted as a component of neutrophils and eosinophils as part of their antibacterial response [240, 241]. Therefore, extracellular DNA represents cellular necrosis and the presence of inhibitors of phagocytosis of apoptotic bodies [242]. The resultant elevated content of DNA contributes to the viscosity of secretions and the inability to clear them. Attempts to reduce viscosity of the pulmonary secretions in CF patients respond best to DNAse [243–245]. Cimmino et al. demonstrated that administration of dornase alpha (a recombinant DNAse) between 4 weeks and 12 months after surgery was associated with improved nasal symptoms and rhinoscopic findings [244]. Furthermore, a double-blind placebo-controlled study found that dornase alpha improved the quality of life outcome measures in CF patients who had previously undergone ESS [246].
Treatment of CRS in CF involves a multidisciplinary approach as the physiology of the upper and lower airways is similar. Surgical treatment for CF typically requires aggressive measures to facilitate gravity-based drainage [247]. Unfortunately, despite the long-term benefits of aggressive surgical therapy, polyp regrowth is common necessitating revisions [248].
Other Forms of Chronic Rhinosinusitis
Granulomatous Chronic Rhinosinusitis
There are three granulomatous diseases that are not uncommonly associated with CRS: granulomatosis with polyangiitis (Wegener’s), Churg–Strauss disease, and sarcoidosis. All cause local inflammatory processes in the sinonasal cavity and upper airways. An astute practitioner should consider them when evaluating a patient for sinonasal complaints.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


