Adult xanthogranuloma presents most commonly as an orange-tan firm solitary nodule with no systemic manifestations. Recently, some cases have been reported in conjunction with lymphoproliferative disorders. Adult reticulohistiocytosis classically presents as red to yellow-red dermal nodules. In the multicentric form, lesions have a predilection for hands and elbows, with a classic coral bead periungual presentation, and are often associated with symmetric erosive arthritis, particularly of the hands and wrists. The presentation and course of Rosai-Dorfman disease, or sinus histiocytosis with massive lymphadenopathy, can vary. The classic presentation is extensive, painless bilateral cervical lymphadenopathy, but some cases have been entirely extranodal.
Key points
- •
In cases of adult xanthogranuloma, workup for lymphoproliferative disorders should be considered, but in general, treatment should be fairly conservative.
- •
Patients with solitary and multicentric reticulohistiocytosis should receive at minimum age-appropriate screening and thorough history and physical examination with aggressive pursuit of focal findings; the multicentric form is typically treated early and aggressively.
- •
Although the classic presentation of Rosai-Dorfman disease is cervical lymphadenopathy, many atypical cases are reported. Treatment must be tailored to the specific disease course.
Adult xanthogranuloma
Overview
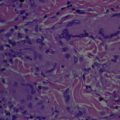
Adult xanthogranuloma is a disease in which lesions are indistinguishable clinically and histopathologically from the lesions of juvenile xanthogranuloma, tan to orange firm nodules ( Fig. 1 ). The natural history of adult xanthogranulomas, however, may be quite different from that of xanthogranulomas in younger patients (see section titled Clinical Presentation below). Histopathologically, early xanthogranulomas consist of large collections of banal-appearing histiocytes in the dermis, sometimes extending into the subcutaneous fat and even the fascia. Later, lesions may show foamy histiocytes, spindled mononuclear cells, Touton giant cells (classically), and also foreign body giant cells, with some eosinophils and lymphocytes also being present. This process is characterized by non-Langerhans cell histiocytes (CD68 + , CD1a − , usually S100 − , and factor XIIIa variable but often positive).
Adult xanthogranuloma strictly defined is considered to be a separate form of histiocytosis that should be distinguished from necrobiotic xanthogranuloma and xanthoma disseminatum (XD).
Etiopathogenesis
The etiopathogenesis of adult xanthogranuloma is poorly understood. Various investigators have posited a role for physical trauma, infection, and malignancy (particularly bloodline tumors) in causing the disease.
Clinical Presentation
Adult xanthogranuloma presents most commonly as a solitary lesion without systemic manifestations. However, extracutaneous manifestations have been rarely reported (see section on Systemic Associations below). It is difficult to know for certain what the natural course of the disease is because many solitary lesions are excised for cosmetic reasons, but in adults with multiple lesions, spontaneous resolution has been noted in about half of the patients. Cases that present with many lesions are perhaps more concerning for association with lymphoproliferative disease; this is discussed further below.
Systemic Associations
Adult xanthogranuloma is primarily a disease of the skin without systemic manifestations. Associations with trauma (because of a few cases in which lesions arose shortly postoperatively), infection, and malignancy have been postulated, but with little to no understanding of a causal mechanism other than perhaps an inflammatory milieu leading to activation of the histiocytes.
Recently, multiple cases of adult xanthogranuloma have been reported in association with lymphoproliferative disorders, including essential thrombocytosis, chronic lymphocytic leukemia, large B cell lymphoma, and monoclonal gammopathy. In all these cases, the presentation was described as multiple xanthogranulomas, disseminated xanthogranulomas, or eruptive xanthogranulomas. In other reports, multiple xanthogranulomas have been described as possibly being associated with a benign course. Although it is not yet entirely clear whether a more acute and widespread presentation is more worrisome for lymphoproliferative disease or whether there is simply variation in terminology, these reports do suggest that an underlying lymphoproliferative disease be considered in patients with eruptive or multiple lesions.
Extracutaneous manifestations of the disease itself, including cervical spine, intracardiac, and periocular lesion location (with the last being possibly associated with adult-onset asthma), have been rarely reported. Although rare, when periocular xanthogranulomas are associated with adult-onset asthma, they are more likely to be associated with lymphadenopathy and/or lymphoproliferative disease.
Evaluation and Management
As the differential diagnosis for adult xanthogranuloma is large, all patients with this suspected diagnosis should receive a thorough history including full review of systems, a full physical examination, and skin biopsy. Eruptive xanthogranulomas in an adult should prompt workup for lymphoproliferative disorder. Periocular xanthogranulomas require an ophthalmologic evaluation in addition to evaluation for asthma, lymphadenopathy, and lymphoproliferative disease. Other workup can likely be pursued as indicated based on any focal examination findings or symptoms.
Periorbital lesions should be followed up and treated locally (eg, topical corticosteroid or surgery) if appropriate, because they may have ophthalmologic complications. Otherwise, use of chemotherapeutic agents, radiation, systemic corticosteroid, and cyclosporine have been reported, but data on efficacy are minimal. As many cases may be indolent and/or self-resolving, management should not be overly aggressive such that harm outweighs benefit.
Adult xanthogranuloma
Overview
Adult xanthogranuloma is a disease in which lesions are indistinguishable clinically and histopathologically from the lesions of juvenile xanthogranuloma, tan to orange firm nodules ( Fig. 1 ). The natural history of adult xanthogranulomas, however, may be quite different from that of xanthogranulomas in younger patients (see section titled Clinical Presentation below). Histopathologically, early xanthogranulomas consist of large collections of banal-appearing histiocytes in the dermis, sometimes extending into the subcutaneous fat and even the fascia. Later, lesions may show foamy histiocytes, spindled mononuclear cells, Touton giant cells (classically), and also foreign body giant cells, with some eosinophils and lymphocytes also being present. This process is characterized by non-Langerhans cell histiocytes (CD68 + , CD1a − , usually S100 − , and factor XIIIa variable but often positive).
Adult xanthogranuloma strictly defined is considered to be a separate form of histiocytosis that should be distinguished from necrobiotic xanthogranuloma and xanthoma disseminatum (XD).
Etiopathogenesis
The etiopathogenesis of adult xanthogranuloma is poorly understood. Various investigators have posited a role for physical trauma, infection, and malignancy (particularly bloodline tumors) in causing the disease.
Clinical Presentation
Adult xanthogranuloma presents most commonly as a solitary lesion without systemic manifestations. However, extracutaneous manifestations have been rarely reported (see section on Systemic Associations below). It is difficult to know for certain what the natural course of the disease is because many solitary lesions are excised for cosmetic reasons, but in adults with multiple lesions, spontaneous resolution has been noted in about half of the patients. Cases that present with many lesions are perhaps more concerning for association with lymphoproliferative disease; this is discussed further below.
Systemic Associations
Adult xanthogranuloma is primarily a disease of the skin without systemic manifestations. Associations with trauma (because of a few cases in which lesions arose shortly postoperatively), infection, and malignancy have been postulated, but with little to no understanding of a causal mechanism other than perhaps an inflammatory milieu leading to activation of the histiocytes.
Recently, multiple cases of adult xanthogranuloma have been reported in association with lymphoproliferative disorders, including essential thrombocytosis, chronic lymphocytic leukemia, large B cell lymphoma, and monoclonal gammopathy. In all these cases, the presentation was described as multiple xanthogranulomas, disseminated xanthogranulomas, or eruptive xanthogranulomas. In other reports, multiple xanthogranulomas have been described as possibly being associated with a benign course. Although it is not yet entirely clear whether a more acute and widespread presentation is more worrisome for lymphoproliferative disease or whether there is simply variation in terminology, these reports do suggest that an underlying lymphoproliferative disease be considered in patients with eruptive or multiple lesions.
Extracutaneous manifestations of the disease itself, including cervical spine, intracardiac, and periocular lesion location (with the last being possibly associated with adult-onset asthma), have been rarely reported. Although rare, when periocular xanthogranulomas are associated with adult-onset asthma, they are more likely to be associated with lymphadenopathy and/or lymphoproliferative disease.
Evaluation and Management
As the differential diagnosis for adult xanthogranuloma is large, all patients with this suspected diagnosis should receive a thorough history including full review of systems, a full physical examination, and skin biopsy. Eruptive xanthogranulomas in an adult should prompt workup for lymphoproliferative disorder. Periocular xanthogranulomas require an ophthalmologic evaluation in addition to evaluation for asthma, lymphadenopathy, and lymphoproliferative disease. Other workup can likely be pursued as indicated based on any focal examination findings or symptoms.
Periorbital lesions should be followed up and treated locally (eg, topical corticosteroid or surgery) if appropriate, because they may have ophthalmologic complications. Otherwise, use of chemotherapeutic agents, radiation, systemic corticosteroid, and cyclosporine have been reported, but data on efficacy are minimal. As many cases may be indolent and/or self-resolving, management should not be overly aggressive such that harm outweighs benefit.
Reticulohistiocytosis
Overview
For the purposes of this article, the definition of reticulohistiocytosis is limited to 2 specific disease manifestations, solitary reticulohistiocytosis or giant cell reticulohistiocytosis (also known as reticulohistiocytoma), and multicentric reticulohistiocytosis. Both these diseases are non-Langerhans histiocytoses limited primarily to adults; this excludes congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease), which is a congenital Langerhans-cell-type histiocytosis.
The histopathologies of solitary reticulohistiocytosis and multicentric reticulohistiocytosis are identical: a dermal collection of lymphocytes and histiocytes as well as scattered plasma cells and eosinophils. Histiocytes characteristically demonstrate a ground glass appearance, that is, copious eosinophilic granular cytoplasm.
Etiopathogenesis
The etiopathogenesis of reticulohistiocytoses, both solitary and multicentric, is not well understood. Associations with other systemic processes is more common in multicentric reticulohistiocytosis and includes autoimmune disease, malignancy, and trauma. Whether these associated conditions are pathophysiologically related to the development of reticulohistiocytomas or are a nonspecific reactive phenomenon is not known.
Clinical Presentation
Solitary reticulohistiocytoma is sometimes said to be a disease of young adults, although the age of onset in one of the larger case series ranged from 2.5 to 74 years. Lesions are classically raised, fairly well-circumscribed, red or yellow-red dermal nodules, ranging from a few millimeters to about 2 cm. Lesions may also be pink, brown, or gray.
Multicentric reticulohistiocytosis is primarily a disease of middle-aged white women, with mean age of diagnosis of 40 to 50 years, although some investigators question whether the disease may be overreported in this group and underreported in others.
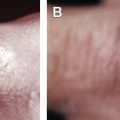
Multicentric reticulohistiocytosis is characterized by widespread papulonodular skin lesions, with a predilection for the hands and elbows, and a characteristic coral bead appearance of periungual lesions ( Figs. 2 and 3 ). This condition is often associated with a symmetric erosive arthritis, particularly of the hands and wrists, and may have other systemic manifestations.
Joint symptoms occur first in nearly 40% of cases, sometimes accompanied by fever, fatigue, and weight loss. Skin symptoms occur first in 30% of cases, and joint and skin symptoms occur simultaneously in the remaining 30% of cases. Dysphagia may also rarely be a presenting symptom because of esophageal involvement.
In one review, hands were most commonly affected by nodules, with the face, arms, trunk, legs, ears, mucosa, and neck also being affected. The classic coral beads sign was present in 27%, palpebral xanthelasmas were present in 17%, and lesions described as vermicular, at the edge of nares, were present in 15%. The course is often relapsing and remitting, with some cases resolving after around 7 years, and 11% to 45% of cases progressing to severe debilitating arthritis.
Other associated signs and symptoms include weight loss (15%), anorexia (4%), dysphagia (3%), pruritus (10%), weakness (10%), cardiac symptoms (9%), myalgia (6%), fever (5%), malaise (5%), and lymphadenopathy (3%).
Systemic Associations
Multicentric reticulohistiocytosis has been reported with a wide array of systemic diseases ( Table 1 ). These diseases include disorders classically defined as autoimmune (primary biliary cirrhosis, systemic sclerosis, systemic lupus erythematosus, dermatomyositis, and Sjögren disease), as well as diabetes, celiac disease, hyperlipidemia, systemic vasculitis, and thyroid disease. This condition has also been associated with many solid organ and lymphoproliferative malignancies, with no particular type of malignancy overrepresented. The efficacy of control of multicentric reticulohistiocytosis via successful treatment of the underlying malignancy is a subject of debate. Finally, multicentric reticulohistiocytosis has been associated with pregnancy, along with a possible increased risk of preeclampsia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


