Adult orbital xanthogranulomatous diseases are rare entities and encompass a group of disorders with varying manifestations that are poorly understood. Taken as a group, there are non-Langerhans histiocytic disorders (type II) that are diagnosed histologically by the presence of foamy histiocytes, Touton giant cells, and varying degrees of fibrosis. Based on the accompanying systemic associations, there are 4 main categories of adult xanthogranulomatous disease: adult-onset xanthogranuloma, adult-onset asthma and periocular xanthogranuloma, necrobiotic xanthogranuloma, and Erdheim–Chester disease. Herein, we discuss the etiopathogenesis, systemic associations, methods of diagnosis, and treatment options for these disorders.
Key points
- •
Taken as a whole, the adult orbital xanthogranulomatous disorders are rare.
- •
Recognition of these lesions are important owing to the occasionally clinically severe systemic associations seen in the different entities.
- •
Treatment options vary with no current consensus as to the most optimum therapeutic course.
- •
Prognosis of patients afflicted with these disorders depends primarily on the presence and severity of any accompanying systemic manifestations.
Introduction
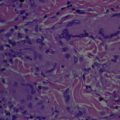
Adult orbital xanthogranulomatous disease (AOXGD) comprises a heterogeneous group of rare orbital and ocular adnexal disorders that are classified as class II non-Langerhans histiocytic proliferations. Based primarily on the degree of systemic involvement, this disease can be classified into 4 subtypes: adult-onset xanthogranuloma, necrobiotic xanthogranuloma (NXG), Erdheim–Chester disease (ECD), and adult-onset asthma and periocular xanthogranuloma (AAPOX; Table 1 ). Although some differences are present histologically, each of these entities is characterized by infiltration of foamy histiocytes and Touton-type giant cells, both of which are often negative for S100 protein and CD1a. Clinically, this infiltration, along with accompanying lymphocytes and varying degrees of fibrosis, can replace the normal lacrimal gland architecture, causing mass effects and loss of tear production. This review discusses the etiopathogenesis of these lesions, along with clinicopathologic presentation and associated systemic manifestations. We conclude with a discussion about current evaluation and management in patients found to have this disease.
| Disease Type | Clinical Features and Systemic Associations | Prognosis | Therapeutic Modalities | Level of Evidence |
|---|---|---|---|---|
| Adult onset | Localized to the eye (anterior orbit) | Good; limited to eye | Surgical debulking | C |
| Adult onset with asthma | Anterior orbit; asthma and lymphadenopathy | Good; rarely associated with systemic findings | Surgery often successful; corticosteroids | C |
| Erdheim–Chester disease | Intraconal; long bone sclerolytic destruction; retroperitoneal fibrosis; cardiac involvement | Poor; mortality rate is increased (∼66%) | Corticosteroids and immunosuppressive agents | C |
| Necrobiotic xanthogranuloma | Anterior orbit ulceration; multiple myeloma, lymphoma, paraproteinemia | Poor; systemic lymphoproliferative disorders | Combination of corticosteroid with immunosuppressive agent; surgical debulking is not recommended | C |
Introduction
Adult orbital xanthogranulomatous disease (AOXGD) comprises a heterogeneous group of rare orbital and ocular adnexal disorders that are classified as class II non-Langerhans histiocytic proliferations. Based primarily on the degree of systemic involvement, this disease can be classified into 4 subtypes: adult-onset xanthogranuloma, necrobiotic xanthogranuloma (NXG), Erdheim–Chester disease (ECD), and adult-onset asthma and periocular xanthogranuloma (AAPOX; Table 1 ). Although some differences are present histologically, each of these entities is characterized by infiltration of foamy histiocytes and Touton-type giant cells, both of which are often negative for S100 protein and CD1a. Clinically, this infiltration, along with accompanying lymphocytes and varying degrees of fibrosis, can replace the normal lacrimal gland architecture, causing mass effects and loss of tear production. This review discusses the etiopathogenesis of these lesions, along with clinicopathologic presentation and associated systemic manifestations. We conclude with a discussion about current evaluation and management in patients found to have this disease.
| Disease Type | Clinical Features and Systemic Associations | Prognosis | Therapeutic Modalities | Level of Evidence |
|---|---|---|---|---|
| Adult onset | Localized to the eye (anterior orbit) | Good; limited to eye | Surgical debulking | C |
| Adult onset with asthma | Anterior orbit; asthma and lymphadenopathy | Good; rarely associated with systemic findings | Surgery often successful; corticosteroids | C |
| Erdheim–Chester disease | Intraconal; long bone sclerolytic destruction; retroperitoneal fibrosis; cardiac involvement | Poor; mortality rate is increased (∼66%) | Corticosteroids and immunosuppressive agents | C |
| Necrobiotic xanthogranuloma | Anterior orbit ulceration; multiple myeloma, lymphoma, paraproteinemia | Poor; systemic lymphoproliferative disorders | Combination of corticosteroid with immunosuppressive agent; surgical debulking is not recommended | C |
Etiopathogenesis
Orbital xanthogranulomatous disease is one of the non-Langerhans (type II) histiocytosis characterized by localized or systemic proliferation of histiocytes. Histiocytes are produced from bone marrow stem cells and mature into monocytes, when they then enter into either the mononuclear–phagocytic system or the dendritic cell system. These 2 groups are composed of unique cell types. The mononuclear–phagocytic system consists of phagocytic monocytes and free and fixed tissue macrophages. The dendritic cell includes the follicular cells of the lymph nodes and the Langerhans cells of the skin, which both serve as antigen-presenting cells. The current thinking is that xanthogranulomas develop secondary to a reactive proliferation of the free tissue macrophages. Although the etiopathogenesis of these proliferations is not known currently, a number of proposed mechanisms have been reported.
In 1971, Balfour and colleagues, presented a case of an 11-week-old male infant with a xanthogranuloma of the salivary gland associated with cytomegalovirus infection. More recently, in 2001, Vasconcelos and colleagues confirmed immunohistochemically the presence of immunolabeling for cytomegalovirus antigens in the histiocytes of an oral juvenile xanthogranuloma. These findings suggest the possibility that xanthogranulomatous diseases may be a virus-induced process. In addition, other patients with juvenile xanthogranulomas demonstrated chromosomal instability in lesional tissue and peripheral blood cells. It is theorized that chromosomal abnormalities constitute either a basic genetic defect or these aberrations may simply serve as evidence of a cellular response to an environmental agent (eg, virus), which would then initiate the proliferation of histiocytes.
With regard to specific entities, the underlying mechanism for the pathogenesis of AAPOX is understood poorly, but recent research suggests a poorly understood systemic immunologic derangement with concurrent bronchiolar and ocular adnexal dysfunction. In addition, because the association of NXG with paraproteinemia is well-known, this fact has led to a number of hypotheses regarding the pathogenesis of this variant of AOXGD. The paraproteinemia present is an immunoglobulin (Ig)G monoclonal gammopathy that is found in at least 80% of cases of NXG. The prevailing thought is that the paraprotein serves as either the primary inciting factor or it may act as a cofactor that facilitates in eliciting the characteristic giant cell granulomatous reaction. To date, however, the precise pathogenesis of NXG and the other adult orbital xanthogranulomas is unknown.
Clinical presentation
Adult-onset xanthogranuloma is rare and as a group it affects patients from 17 to 85 years of age with no significant gender preference (73 male vs 64 female cases). Adult-onset xanthogranuloma is an isolated xanthogranulomatous lesion without significant systemic involvement. It is the least common entity among AOXGDs, and affects patients ranging from 38 to 79 years with no significant gender preference. The significance of recognizing this entity is that it is often self-limited and does not require aggressive treatment, in contradistinction to the other subtypes of AOXGDs, which can sometimes cause significant consequences.
NXG is characterized by the presence of subcutaneous skin lesions that tend to ulcerate and become fibrotic. The most common clinical manifestation is an indurated papule, nodule, or plaque that can be violaceous, red-orange, or xanthomatous (yellow) in color. Papules usually progressively enlarge to bulky nodules and plaques with scarring, atrophy, and telangiectasias. Ulcerations, which may be extensive, are seen in more than 40% of patients. The size of the plaques can range from 0.3 to 25 cm. The most common site of involvement is the periorbital region (>80% of cases with other parts of the face, trunk, and extremities). Periorbital lesions may involve single or multiple eyelids and may be unilateral or bilateral. This entity often affects adults 20 to 85 years of age with no significant gender preference (32 for male and 40 for female).
ECD is an idiopathic condition of lymphohistiocytic infiltration in the orbit as well as internal organs, including the heart, lungs, retroperitoneum, bones, and other tissues. These patients typically present with bone pain and characteristic lesions are seen radiographically on further evaluation, which are ultimately what lead clinicians to arrive at the proper diagnosis. Other systemic symptoms leading to further workup include fever, weakness, weight loss, and night sweats. Whereas skeletal manifestations are pathognomonic, cutaneous manifestations that may elevate the clinical suspicion include orbital disease with exophthalmos and xanthoma-like lesions of the eyelids. Like AAPOX, it presents preferentially in male (male to female: 2:1) and affects patients ranging from 7 to 84 years of age. This condition is often fatal, with death owing to cardiomyopathy, severe lung disease, or chronic renal failure. In addition, bone involvement is common, with frequent critical consequences despite aggressive therapies. Because of its frequent and significant systemic involvement, bilateral diffuse orbital masses should alert the clinician to the possibility of this serious systemic disease.
AAPOX is rare, with fewer than 30 reported cases in the literature and a newly diagnosed case at our institution. It often presents with bilateral yellow-orange, elevated, indurated, and nonulcerated xanthomatous eyelids and/or orbital masses. It typically extends into the anterior orbital fat, and sometimes involves the extraocular muscles and/or the lacrimal gland(s). AAPOX is most often seen in adult patients ranging from 22 to 74 years old, with twice as many males being affected as females. Most patients experience adult-onset asthma within a few months to a few years of the onset of the orbital lesions. Interestingly, asthma has also been reported with some frequency in patients with sinus histiocytosis and massive lymphadenopathy. Even when asthmatic symptoms are severe enough to require systemic corticosteroids and inhalation therapy, a chest radiograph may be negative.
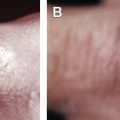
Because the eyelids usually remain intact and these xanthogranulomatous processes typically do not reach the deep orbital and perioptic nerve connective tissues, visual acuity may not be affected and ocular motility is generally well-preserved, unless the extraocular muscles are involved. Only rarely are the corneas exposed to the infiltrate, but in the rare cases where lacrimal gland involvement causes punctuate corneal epitheliopathy, the patient may suffer from a dry eye condition. However, the eyelid and periocular infiltration can be very extensive and cause significant mass effect that prevents the patient from seeing properly ( Fig. 1 ).