Acantholytic Disorders of the Skin: Introduction
|
Darier Disease
Darier disease (DD) (OMIM #124200) is an autosomal dominant disease affecting both sexes and all ethnic groups. DD was described independently by Darier and White in 1889 (also known as Darier–White disease or keratosis follicularis).1 Estimates of prevalence range from 1 in 30,000 (Northeast England, Scotland, Slovenia)2–4 to 1 in 100,000 (Denmark).5 Penetrance is complete, but spontaneous mutations are frequent.
The gene for DD was mapped by linkage analysis to chromosome region 12q23-24 in 19936 and ATP2A2 was identified as the defective gene in 1999.7 ATP2A2 encodes sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase (ATPase) isoform 2 (SERCA2), a calcium pump transporting Ca2+ from the cytosol to the lumen of the endoplasmic reticulum (ER).8–11 DD is caused by mutations inactivating one ATP2A2 allele.
ATP2A2 spans 76 kilobases (kb), is organized in 21 exons, and encodes a 4.4-kb transcript, which is alternatively spliced into three isoforms: (1) SERCA2a, (2) SERCA2b, and (3) SERCA2c.12,13 SERCA2a is expressed in the slow-twitch skeletal muscles and cardiac muscle, unaffected in DD.14,15 SERCA2b and SERCA2c are expressed ubiquitously, but SERCA2b is the major isoform detected in the human epidermis.16 Mutations specific for SERCA2b are sufficient to cause DD (despite the presence of functional SERCA2a), confirming the important role of SERCA2b in epidermis.17,18 Most tissues may compensate for deficiencies in SERCA2 by mechanisms such as SERCA3, which is not expressed in keratinocytes.16
SERCA2 pumps belong to the P-type Ca2+ ATPase family. The pumps catalyze the hydrolysis of adenosine triphosphate (ATP) coupled with the translocation of two Ca2+ ions from the cytosol to the ER lumen, where Ca2+ is stored at high concentrations. SERCA pumps comprise three cytoplasmic domains [(1) the actuator, (2) phosphorylation, and (3) ATP-binding domains] linked to a transmembrane domain with ten transmembrane helices that contain the two Ca2+-binding sites. After binding of cytosolic Ca2+ ions and phosphorylation, the pump undergoes conformational changes and releases Ca2+ into the ER lumen.19
More than 140 pathogenic ATP2A2 mutations have been reported, including missense mutations (50%), frameshift mutations (23%), and in-frame deletions or insertions (8%). No consistent correlation has been demonstrated between genotype and phenotype, but missense mutations may be associated with more severe forms.7,17,18,20–33 The considerable phenotypic variability suggests that other genetic or environmental factors modify the phenotype.
Although the etiology has been explained, the pathogenesis of DD is less clear. High concentrations of Ca2+ are required for normal keratinocyte intercellular adhesion and differentiation. Normally, the epidermis displays an increasing epidermal Ca2+ gradient from the basal to the superficial layers, but in DD this gradient is disturbed. The level of Ca2+ is reduced in basal cells from both affected and unaffected skin.34
The earliest ultrastructural change is breakdown of desmosomes with aggregation of keratin filaments around the cell nucleus.35 Immunohistological studies of acantholytic cells reveal internalization of desmosomal components.36–38 The dyskeratotic cells in the epidermis (grains, corps ronds) are formed through apoptosis, which appears to be triggered by the loss of adhesion.39 The expression of antiapoptotic proteins in the Bcl-2 gene family is reduced in DD, possibly as a secondary phenomenon, but an alteration in Bcl-2 proteins might also contribute to apoptosis.39–42
SERCA pumps replenish the ER Ca2+ pool from cytosolic Ca2+, but mutations disrupting critical functional domains reduce activity of the pump in DD. A reduced ER Ca2+ pool may impair processing of desmosomal proteins by affecting the function of molecular chaperones such as calreticulin and calnexin that prevent protein misfolding and are involved in posttranslational modification.43 Intracellular signaling through release of ER Ca2+ regulates trafficking of proteins to the cell membrane, including those required for the assembly of desmosomes and adherens junctions.44 The trafficking of desmoplakin to the cell membrane is impaired in cultured DD keratinocytes.45 Variations in cellular Ca2+ concentrations are also likely to have an effect on the expression of Ca2+-dependent genes involved in keratinocyte differentiation and adhesion.
The epidermis is papillomatous and differentiation is abnormal, with the expression of hyperproliferative keratins and premature expression of cornified envelope precursors such as involucrin.34,38,46–48 Alterations in ATP-mediated signaling may contribute to dyskeratosis and hyperproliferation.8,34 Extracellular Ca2+ binds to purinergic ATP receptors that transmit calcium signals into the cytosol. Activation of the G-protein-coupled ATP receptor (P2Y) generates inositol 1,4,5-tris-phosphate (IP3), a calcium-signaling messenger (Fig. 51-1). Binding of IP3 to surface receptors on the ER and Golgi apparatus triggers release of Ca2+ stores and a rise in cytosolic Ca2+ which activates a further influx of extracellular Ca2+ through plasma membrane Ca2+ channels (store-operated Ca2+ entry), including transient receptor potential canonical1 (TRPC1). In DD, depleted ER Ca2+ appears to upregulate TRPC1 and enhances influx of Ca2+.8,49
Figure 51-1
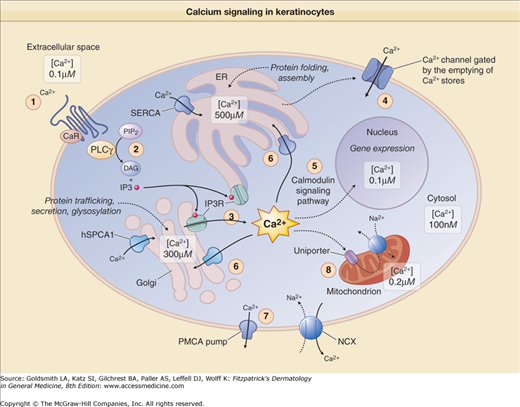
A simplified representation of Ca2+ signaling in keratinocytes. 1. Ca2+ binding to its plasma membrane receptor (CaR) activates phospholipase Cγ (PLCγ). 2. This causes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-tris-phosphate (IP3) and diacylglycerol (DAG). 3. IP3 binds to its receptor (IP3R) at the surface of the endoplasmic reticulum (ER) and Golgi apparatus, which causes the depletion of intracellular stores and induces an increase in intracellular Ca2+ levels. 4. This increase triggers the opening of Ca2+ release-activated channels in the plasma membrane, which leads to a sustained increase in Ca2+ intracellular levels. 5. Ca2+ binds to calmodulin; this activates calcineurin and calmodulin-dependent protein kinases, which regulate gene transcription through phosphorylation/dephosphorylation of transcription factors. 6. Active Ca2+ transport by the different sarco/endoplasmic reticulum calcium adenosine triphosphatase (ATPase) pumps (SERCA1 to SERCA3) and human secretory pathway Ca2+/Mn2+–ATPase (SPCA1) pump is essential to replenish ER and Golgi Ca2+ stores, respectively. SPCA1 is also required for Mn2+ influx into the Golgi apparatus (not indicated here). 7. Ca2+ efflux to the extracellular space involves plasma membrane Ca2+–ATPases (PMCA) and Na+/Ca2+ exchangers (NCX). 8. Mitochondria take up Ca2+ released from the internal stores during Ca2+ signaling via the Ca2+ uniporter and return it to the cytosol through an NCX. Thus, Ca2+ homeostasis requires differential Ca2+ concentrations in the cytosol, the sarco/endoplasmic reticulum, the Golgi apparatus, the mitochondria, and the nucleus of the cell. The largest store of cellular Ca2+ is located in the ER lumen and in Ca2+-binding proteins. Ca2+ signaling is highly regulated and generated by influx through Ca2+ receptors, release from internal stores (ER, Golgi apparatus, mitochondrion), and sequestration by Ca2+ pumps (SERCAs, SPCA1) and Ca2+ exchangers. mNCX = mitochondria Na2+/Ca2+ exchanger.
Elevated cytosolic Ca2+ may drive keratinocyte proliferation by activation of Ca2+-dependent messenger systems that regulate cell division and differentiation.49 Mice with reduced SERCA2 develop papillomas and SCCs.50 SERCA2 haploinsufficiency in cultured mouse keratinocytes is linked to upregulation of proliferation but downregulation of differentiation.51 SCC has been reported rarely in DD, sometimes in association with HPV16.52–56
Much of the skin may be able to compensate for the deficiency in SERCA2 by increasing the expression of the normal SERCA2 allele or by upregulating other mechanisms such as the human secretory-pathway Ca2+/Mn2+ ATPase isoform 1 (SPCA1) in the Golgi (see section “Hailey–Hailey Disease”). However, external factors such as ultraviolet B (UVB) irradiation or friction, which are known to exacerbate DD, may disrupt this subtle balance by downregulating ATP2A2 or by increasing the requirement for SERCA2 until the protein reaches a critical level.8,57 This hypothesis is supported by the observation that UVB irradiation and proinflammatory cytokines reduce levels of ATP2A2 mRNA in cultured normal keratinocytes and that retinoids and corticosteroids (used in treatment of DD) prevent this reduction.58 Lithium, another well-known trigger for DD, reduces epidermal expression of SERCA2 in rats.59
(Box 51-1). The first manifestations usually appear between the ages of 6 and 20 years with a peak between 11 and 15 years, but DD may develop in infants or old age.1,60 Symptoms include itch, malodor, and pain. Heat, sweating, friction, and sunlight (UVB) exacerbate the signs that may be noticed for the first time in hot summer months.1,2,61
|
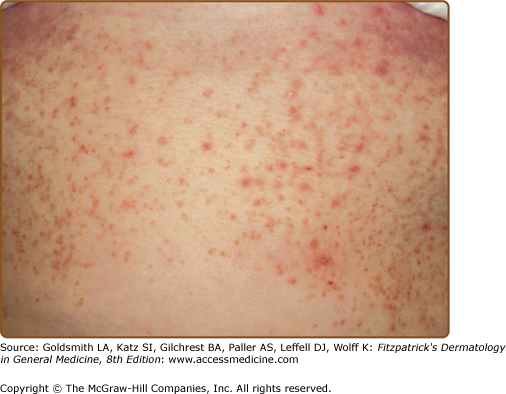
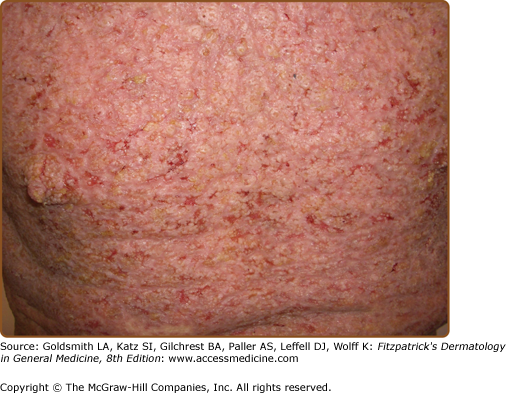
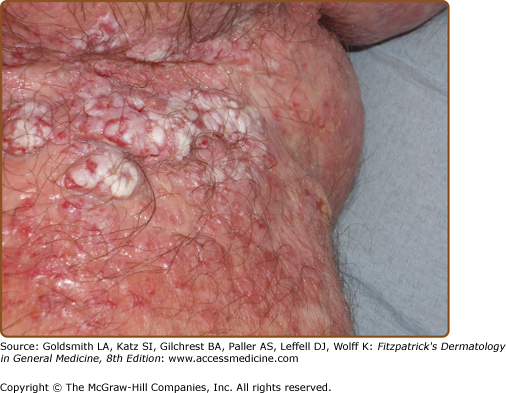
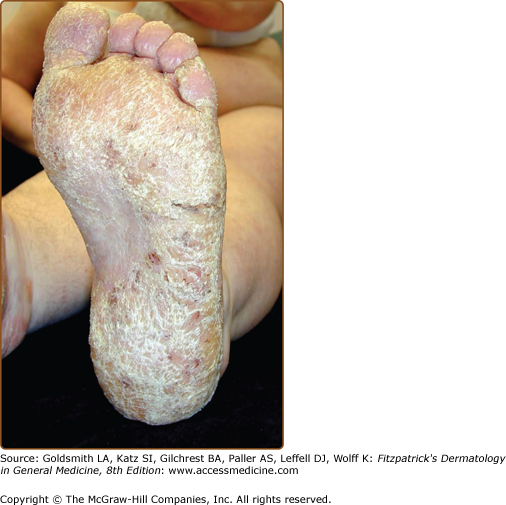
The discrete, greasy, yellowish-brown keratotic papules (only some are perifollicular) have a predilection for seborrheic areas: central chest and upper back, scalp (hair growth is not affected), forehead, neck including supraclavicular fossae, ears, and skin creases (axillae, groins, and perineum) (Figs. 51-2 and 51-3). Papules tend to coalesce into crusted plaques (Fig. 51-4). Foul-smelling, hypertrophic disease in the groin is particularly disabling (Fig. 51-5). In 1889, White noted the “intolerable stench” that accompanies severe disfiguring disease.62 DD may affect schooling, work, and relationships.1,61,63
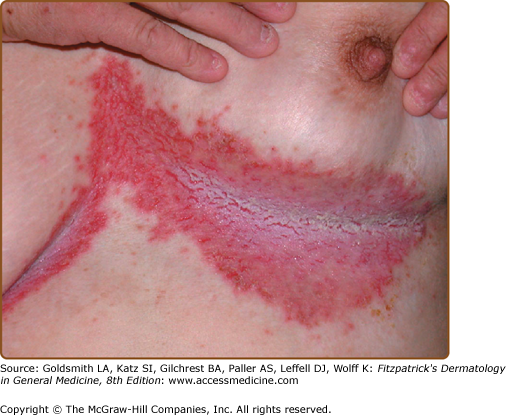
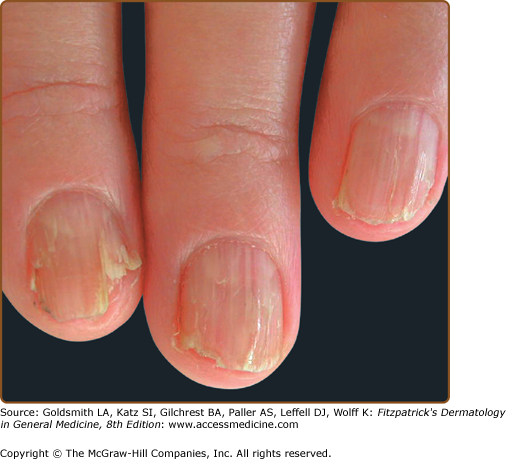
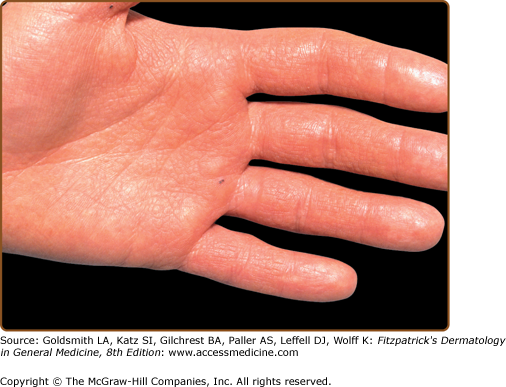
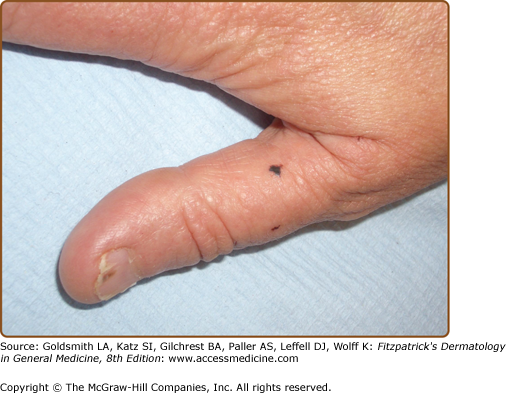
Hands and/or nails are affected in >96% of patients and may show the first signs of disease.1,2 Look for nail fragility, painful longitudinal splits, or distinctive red and white longitudinal bands terminating in V-shaped nicks (Fig. 51-6). Pits or keratotic papules on palms and, sometimes, soles, help to confirm the diagnosis (Fig. 51-7). Many patients (50–70%) have skin-colored, flat-topped papules on the dorsa of hands and/or feet like those of acrokeratosis verruciformis of Hopf (AKV).64 Hemorrhagic macules with jagged margins, possibly linked to trauma, are the least common acral sign and may blister (Fig. 51-8). Hemorrhagic DD has been reported in some kindreds as well as individuals.65–68
Oral,1,69–71 esophageal,72,73 rectal,74 and cervical75 mucosa may be affected. Corneal abnormalities have been recorded.76–78
Variants include painful erosive DD,79 vesiculobullous DD,80 grossly hyperkeratotic plaques (cornifying DD),81–83 nipple hyperkeratosis,84 keratoderma (Fig. 51-9), comedonal DD,33,85,86 freckled “Groveroid” DD87 and, in pigmented skin, guttate leukoderma with confetti-like hypopigmented macules, and/or papules.88,89
Type 1 mosaicism presents with one or more unilateral bands of keratotic papules following Blaschko’s lines. Hands and/or nails may be affected on the same side90,91 (Fig. 51-8). This distribution reflects a postzygotic somatic mutation in ATP2A2 early in embryogenesis.92–94 Theoretically, patients with gonadal mosaicism could transmit generalized disease, but no such cases have been reported. Type 2 mosaicism is rare and characterized by a severely affected linear band of DD, superimposed on generalized disease. A postzygotic mutation causes loss of heterozygosity at the ATP2A2 locus in the more severely affected skin.95
DD has occasionally been reported in association with neuropsychiatric disease including seizures, bipolar disorder, and schizophrenia.1,32,96–98 Lithium, which may be prescribed for bipolar disorder, exacerbates disease, possibly by suppressing levels of epidermal SERCA2.59,99–101 The lifetime prevalence of major depression (30%), suicide attempts (13%), and suicidal thoughts (31%) appears higher than in the general population, highlighting the need for careful assessment.98,102 Reports of cosegregation of DD with bipolar disorder in some families support the existence of a bipolar disorder susceptibility gene in the DD region, but ATP2A2 has been excluded as a common susceptibility gene for bipolar disease.103–105 Learning difficulties reported in some patients may be, at least in part, secondary to social handicap caused by disfigurement. Bone changes, particularly bone cysts, have been reported infrequently.106–108
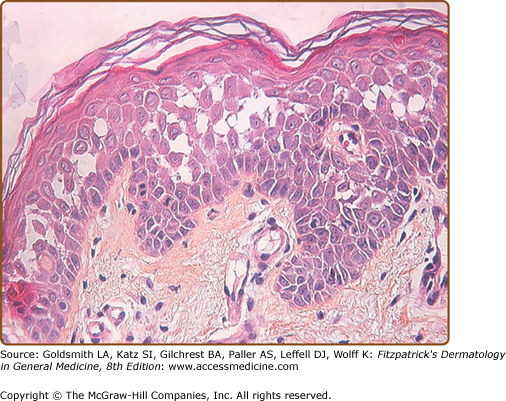
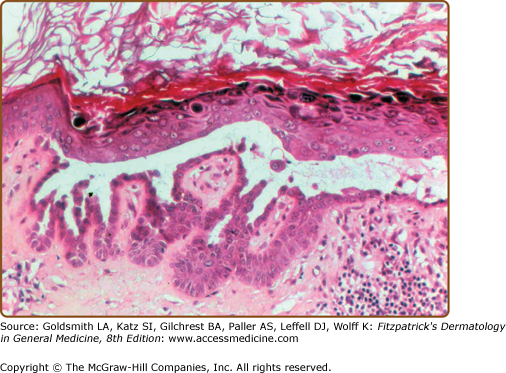
Histologic examination shows downgrowths of narrow cords of keratinocytes, suprabasal acantholysis with suprabasal clefts (lacunae), dyskeratosis (premature and abnormal keratinization), and hyperkeratosis (Fig. 51-10). Apoptosis results in rounded eosinophilic dyskeratotic cells in the epidermis (corps ronds) and flattened parakeratotic cells in the horny layer (grains).39 The warty papules on the backs of the hands show the histology of AKV (see below).
(Box 51-2)
SEBORRHEIC
EROSIVE
VEGETATING FLEXURAL
COMEDONAL
ACRAL
FRECKLED
GENITAL
|
DD may be misdiagnosed as seborrheic dermatitis or acne, particularly in patients without a family history. Look for signs of DD in the hands or nails. Acneiform facial DD may be confused with familial dyskeratotic comedones109,110 or comedo-like acantholytic dyskeratosis.111
Erosive, bullous, or hypertrophic flexural disease simulates Hailey–Hailey disease (HHD), but HHD tends to present later and patients do not have keratotic papules or nail fragility.112 Erosive or hypertrophic DD may also resemble pemphigus vulgaris or vegetans (see Chapter 54), but patients do not have mucosal ulcers and intracellular immunoglobulin (Ig) and complement are not detected in the epidermis. Localized hypertrophic DD may suggest malignancy (Fig. 51-5). Freckled or papulovesicular forms may resemble Grover disease (GD),87 but GD is not familial (see below), or the rare acantholytic variant of Dowling–Degos disease (Galli–Galli disease).113,114
Acral papules resemble plane warts or AKV (which may be a form of DD).64,115,116 Localized papular vulvocrural acantholytic disease may be part of the spectrum of DD or HHD.117–120
Impetiginization and eczematization may complicate the picture and patients have an increased susceptibility to widespread infection with Herpes simplex (eczema herpeticum) (Fig. 51-11) or Herpes zoster.121,122 Blockage of salivary glands has been reported.1,69,80,123 SCC (scalp, scrotal, vulval, thigh, subungual) has been recorded infrequently, sometimes linked to the presence of HPV16.52–56
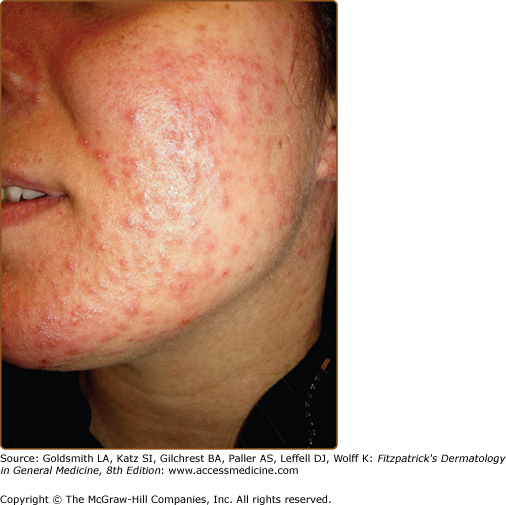
DD pursues a chronic relapsing course. Severity is unpredictable, but in about 30% of patients disease becomes less severe in old age, while in others DD persists or gradually worsens.1,61
(see Table 51-1)
First Line | Second Line | Third Line (Unproven Efficacy) |
|---|---|---|
Discuss how to avoid triggers (heat, sweating, friction) and minimize UVB-induced exacerbations. Emollients containing urea or lactic acid. Soap substitutes and topical antiseptics. Moderate or potent topical corticosteroids with topical antibiotics. Topical retinoids: isotretinoin (0.05%, 0.1%), tretinoin, tazarotene gel, adapalene 0.1% gel. | Oral acitretin 0.25–0.5 mg/kg/day. Takes 3 months to have a maximal effect. Acitretin should be stopped for 2 years before a woman attempts to conceive. Oral isotretinoin 0.5 mg/kg/day. Less effective than acitretin but may be indicated in young women. Isotretinoin should be stopped for 1 month before a woman attempts to conceive. | Topical 5-fluorouracil. Oral cyclosporine for eczematization. Initially 2.5 mg/kg/day. Laser surgery, electrosurgery, or dermabrasion. Photodynamic therapy. Botulinum toxin to reduce sweating in recalcitrant flexural disease. Mammaplasty for severe inframammary disease. |
The physician should take the time to answer questions, explain that treatments may control but not cure DD, and offer genetic counseling if appropriate. It is important to discuss how to reduce the impact of triggers such as heat and sun, and use emollients containing urea or lactic acid to reduce hyperkeratosis. Topical antiseptics (washes, bath additives), antibiotics, and antifungals will help to prevent or treat infection. Herpes simplex causes painful flares that require oral acyclovir.121 Topical corticosteroids in combination with antibiotics reduce inflammation. Limited disease may respond to a topical retinoid, for example, 0.1% tazorotene124,125 or 0.05% isotretinoin,126 prescribed in combination with a topical corticosteroid to reduce irritation. Other topical agents such as adapalene,127,128 5-fluorouracil,129–131 or tacrolimus132 have been reported to be effective in small numbers of cases.
Oral acitretin 0.25–0.5 mg/kg or isotretinoin 0.5 mg/kg reduces hyperkeratosis and malodor, but will take 3 to 4 months to achieve maximal effect.133 Tailor dose to the response and monitor for adverse effects (see Chapter 228). Pregnancy is contraindicated for 2 years after stopping treatment with acitretin and for 1 month after stopping isotretinoin. Oral cyclosporine has been advocated for eczematization134 and in severe vulval disease,135 but controlled trials are lacking.
Approaches for severe hypertrophic or erosive disease include dermabrasion, laser ablation, and photodynamic therapy, but controlled studies are needed to evaluate these approaches.79,133,136,137 Botulinum toxin may control flexural exacerbations by reducing sweating and mammoplasty has been advocated for severe inframammary disease.138,139
Acrokeratosis Verruciformis or Acral Darier Disease
AKV (OMIM #101900) is inherited in an autosomal dominant fashion.115 Sporadic cases have been reported. A missense mutation in ATP2A2, the gene that is affected in DD, was identified in a large British pedigree. The heterozygous Pro602Leu mutation caused complete loss of Ca2+ transport activity.116 Thus, in some instances, AKV and DD are allelic disorders, a conclusion that is entirely consistent with the overlapping clinical features. However, mutations in ATP2A2 were not identified in a Chinese family.140
AKV usually presents at birth or in early childhood. The asymptomatic, skin-colored, flat-topped warty papules are distributed symmetrically on the dorsum of the hands and feet (Fig. 51-12). Papules may also develop on knees, elbows, and extensor aspects of legs and forearms.64 As in acral DD, punctate keratoses and pits may be present on palms and soles, palmar skin may be thickened, and patients may have subungual hyperkeratosis, longitudinal striations, splits, and V-shaped nicks at the free margin of the nail plates. A linear variant with persistent localized unilateral lesions has been reported in two unrelated Saudi patients.141
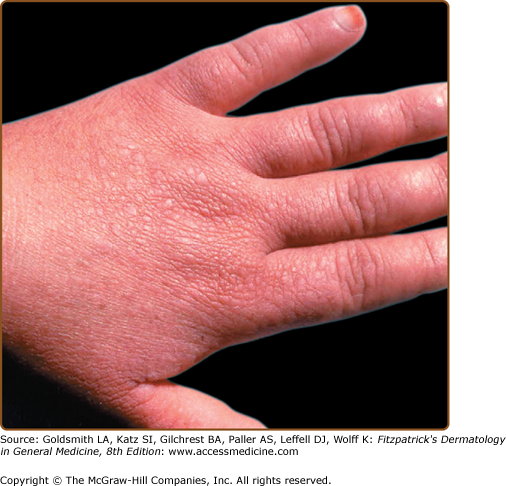
The classic histopathologic findings are hyperkeratosis, hypergranulosis, and acanthosis with papillomatosis. The spiky elevations of the epidermis are said to resemble “church spires” (Fig. 51-13). Epidermis is neither dyskeratotic nor acantholytic.
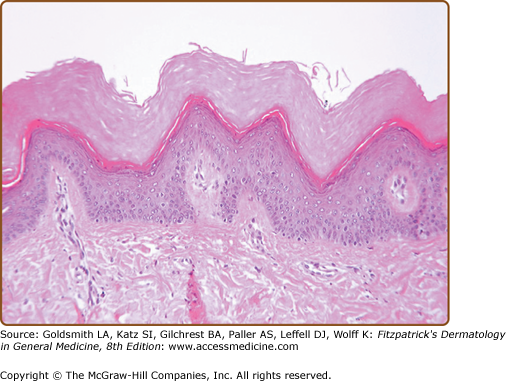
Acral DD may be indistinguishable from AKV, particularly in childhood, when other features of DD may not be apparent. AKV may resemble plane warts, stucco keratoses, or seborrheic warts, but the family history, symmetrical distribution, and nail changes will suggest the diagnosis.
None may be needed. Topical retinoids may flatten lesions. Destruction by cryosurgery, shave, curettage, or laser surgery can be effective. Oral acitretin has been helpful.142
Hailey–Hailey Disease
HHD (OMIM #169600), also known as familial benign chronic pemphigus, was described by the Hailey brothers in 1939. HHD has an incidence of at least 1 in 50,000, but prevalence may be higher as misdiagnosis is frequent.112
The discovery of the crucial role of SERCA2 in DD raised the possibility that defects in another calcium pump could underlie acantholysis in HHD. The defective gene, ATP2C1, was identified in chromosome region 3q21-24. ATP2C1 encodes an ATP-powered calcium channel pump on the Golgi membrane, the human secretory-pathway Ca2+/Mn2+ ATPase isoform 1 (SPCA1).143–145 SPCA1 belongs to the family of P-type cation transport ATPases. HHD is dominantly inherited with complete penetrance and is caused by mutations inactivating one ATP2C1 allele.
The gene spans approximately 30 kb on 3q21 and comprises 28 exons encoding a 4.5-kb transcript. The predicted protein is approximately 115 kDa. Alternative splicing of ATP2C1 primary transcripts in keratinocytes leads to four splice variants, ATP2C1a to ATP2C1d, which differ by different splicing of exon 27 and/or 28. ATP2C1d is the largest variant, containing exons 27 and 28 in their entirety. The structure of SPCA is similar to that of SERCA (see section “Darier Disease”), but the SPCA-binding site only transports a single Ca2+ or Mn2+ ion into the Golgi lumen.19,144,146,147
Around 100 mutations have been reported in HHD scattered across the ATP2C1 gene.143,144,148–161 No correlations have been found between genotype and phenotype; clinical features vary.149 Mutations predict marked reduction of SPCA1 or cause changes in highly conserved domains that are critical for function.143,145,153,162–165 Haploinsufficiency appears to be the mechanism for dominant inheritance, but it is not clear how loss of one functional ATP2C1 allele causes acantholysis.
Ultrastructural studies of acantholytic cells reveal desmosomal breakdown with retraction of keratin filaments from desmosomal plaques to form perinuclear aggregates.166,167 Desmosomal components, E-cadherin, and connexins are internalized in acantholytic cells and the expression of keratins is abnormal in lesional skin.34,36–38,168–172 The abnormality in cell adhesion may be revealed in normal-looking skin in patients with HHD using suction.173
Immunohistochemical studies suggest that SPCA1 is localized to the basal layer of normal epidermis.174 Total [Ca2+] in the epidermal granular layer is reduced and the normal epidermal Ca2+ gradient attenuated in both affected and unaffected skin.143,163 Golgi Ca2+ stores are reduced, Ca2+ signaling is abnormal and the normal upregulation of transcription of ATP2C1 mRNA by Ca2+-stimulation is suppressed in HHD keratinocytes.143,145,163,175–177 Elevated cytosolic Ca2+ levels could influence gene expression or alter posttranslational modification of target proteins. Alternatively, low Ca2+ or Mn2+ concentrations in the Golgi lumen could impair posttranslational modifications (proteolytic processing, glycosylation, trafficking, or sorting) of proteins important in epidermal cell-to-cell adhesion.
Cultured keratinocytes from involved skin display altered patterns of calcium metabolism and reduced proliferative capacity. Increased oxidative stress in HHD keratinocytes may lead to reduced expression of proteins involved in regulation of the balance between proliferation and differentiation such as Itch and Notch1.161,178 Reorganization of actin is defective, cellular ATP decreased, and synthesis of involucrin is reduced.161,179–181 Mice deficient for SPCA1 develop squamous cell papillomas and carcinomas similar to those seen in SERCA2 deficient mice (see section “Darier Disease”).176
Although ATP2C1 mRNA is expressed ubiquitously, HHD is limited to the skin. Keratinocytes may be more sensitive to levels of SPCA1 than other cells because, unlike most other cells, the Golgi in keratinocytes lack SERCA to compensate for deficient SPCA1.147 UVB irradiation and proinflammatory cytokines reduce expression of ATP2C1 mRNA in cultured normal keratinocytes, but suppression is inhibited by retinoids, corticosteroids, cyclosporine, tacrolimus, and vitamin D3.58 External factors (UVB, sweating, friction) may reduce the amount of SPCA1 to a critical level leading to the expression of disease.147
(Box 51-3)
Consider the possibility of HHD in any young adult with chronic “eczema” at sites of friction such as the neckline, axilla, or groins. The diagnosis is often missed. Look for:
|
HHD usually presents between the second and fourth decades, predominantly at sites of friction (neck, axillae, inframammary, groin, perineum).112 The diagnosis is often delayed because HHD simulates common dermatoses such as eczema, tinea, and impetigo.112 Itch, pain, and malodor are common complaints.
Signs include crusted weeping erosions, vesicopustules, expanding annular plaques with peripheral scaly borders and vegetating plaques with fissures (rhagades). Postinflammatory hyperpigmentation is frequent (Figs. 51-14 and 51-15). Some patients have longitudinal white lines on the fingernails and these can help to confirm the diagnosis112,182,183 (Fig. 51-16). Disease may be limited to one or two sites, more widespread or rarely generalized with erythroderma,184–186 but even mild disease reduces quality of life.63,187 Painful malodorous inguinal or perineal disease is particularly disabling. HHD koebnerizes into inflammatory dermatoses and has been exacerbated (or diagnosed) after triggers such as contact dermatitis, removal of adherent patch tests, UV irradiation, cutaneous infections, and scabies infestation.173,186,188–196 Generally, HHD does not involve mucosa. Rare instances of conjunctival, oral, esophageal, or vaginal involvement may have been initiated by trauma or infection.197–203
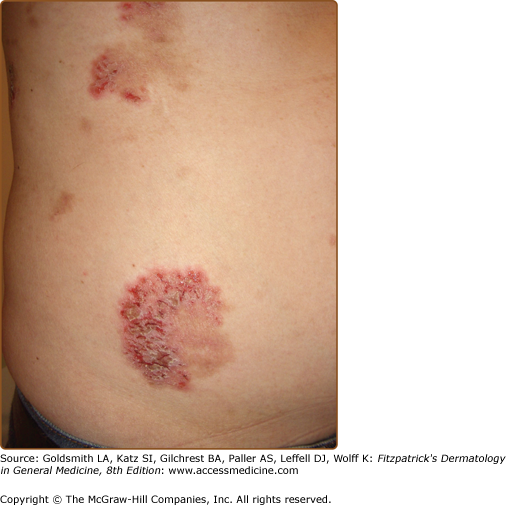
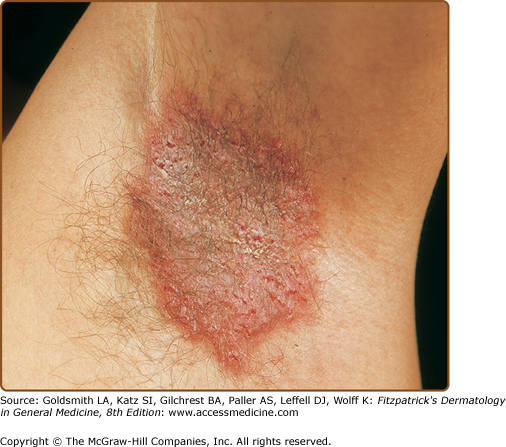
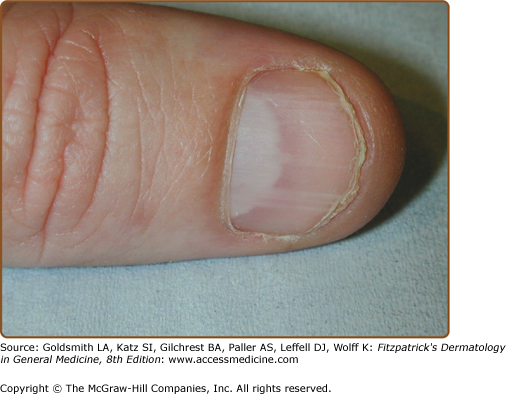
In Type 1 mosaicism, a postzygotic mutation in ATP2C1 manifests as one or more localized streaks of HHD along Blaschko’s lines.204 Type 2 mosaicism has been confirmed in a patient with severe linear involvement superimposed on symmetrical HHD. In the more severely affected skin, a postzygotic mutation had caused loss of heterozygosity at the ATP2C1 locus.205
Involved skin displays widespread partial loss of cohesion (keratinocytes may still be linked together by adherens junctions206) between suprabasal keratinocytes with an appearance likened to a dilapidated brick wall (Fig. 51-17). Clusters of loosely coherent cells float in suprabasal clefts or bullae. Dyskeratosis, when present, is usually mild, but changes may resemble those in DD.