Figure 18.1
A man with a blistering rash
A skin biopsy was taken which showed a subepidermal blister with neutrophils and occasional eosinophils. PAS staining showed that the basement membrane was in the floor of the blister. Immunohistochemistry showed C3 was deposited along the dermoepidermal junction and around the appendages and fibrinogen in the floor of the blister.
He was commenced on Prednisone 60 mg daily, but despite this, new blisters continued to develop. These occurred generally around sites of trauma and old blisters healed with scarring and milia.
Based on the case description and photographs, what is your preferred diagnosis?
1.
Dystrophic Epidermolysis Bullosa
2.
Epidermolysis Bullosa Acquisita
3.
Bullous Pemphigoid
4.
Bullous Systemic Lupus Erythematosus
His prednisone dose was increased to a maximum of 280 mg a day and trials of Dapsone 200 mg daily, Tetracycline 1.5 gm daily, Doxycycline 200 mg daily and Azathioprine at 200 mg daily were used with little improvement in an attempt to control the disease.
A repeated skin biopsy showed a subepidermal blister with lymphocytic and eosinophilic inflammation and positive IgG and C3 along the split on direct immunofluorescence (IF). Indirect IF on human salt-split skin was positive in a strong linear band at the floor of the split. Serum for Western immunoblot testing confirmed autoantibodies to type VII collagen.
Blistering continued despite trials of Colchicine, Cyclosporin A, intravenous immunoglobulin, photophoresis, Sulfasalazine, Mycophenylate and Methotrexate. He developed progressive dysphagia due to esophageal strictures, which resulted in profound weight loss and nutritional deficiencies [1].
The decision was made to trial Rituximab, which was given in four infusions over 3 months at a dose of 375 mg per meter squared of body surface area.
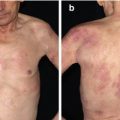
Within 2 months of having the first dose of Rituximab, the amount of new blistering lesions dramatically diminished and he was weaned off Prednisone. Four months after the first infusion, his dysphagia had markedly improved and with an increased oral intake he gained 12 kg in weight (see Fig. 18.2a Pre-Rituximab and 2B Post-Rituximab).
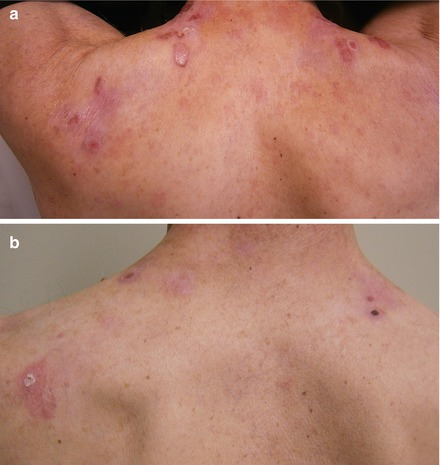
Figure 18.2
(a) EBA pre-Rituximab, (b) EBA post-Rituximab
Diagnosis
Epidermolysis Bullosa Acquisita
Discussion
Epidermolysis Bullosa Acquistia (EBA) is a rare, acquired and chronic autoimmune blistering disease characterized by IgG autoantibodies against type VII collagen resulting in subepidermal blistering with minimal trauma to the skin [2]. EBA presents with generalized blistering and subsequent erosions with associated pruritus, erythema, skin fragility (positive Nikolsky’s sign) and healing with scarring and formation of milia. Type VII collagen is a major component of the anchoring fibrils in the basement membrane zone, which help maintain the adhesion of the dermis to the epidermis. It is the same protein that is affected in hereditary Dystrophic Epidermolysis Bullosa (DEB), which EBA resembles clinically and from which EBA originally derived its name.
DEB is a genetic condition with a defect in the CO7A1 gene that codes for type VII collagen [3]. The recessive and most severe form of DEB usually presents early on in life and is associated with premature mortality [4]. Treatment consists of protective and therapeutic dressings, use of topical preparations and prevention and treatment of complications of the disease. Translational therapies, including gene, cell and protein therapies are in the development phases [5].
EBA can also be clinically indistinguishable from Bullous Pemphigoid (BP), which is the most common autoimmune blistering disease in the older generation. BP is characterized by autoantibodies against the BP180 and BP 230 antigens, which are components of the hemidesmosome adhesion complex [6]. BP normally presents with spontaneous tense blistering, intense pruritus and is not usually associated with skin fragility (negative Nikolsky’s sign), trauma induced blisters, scarring or milia. Histologically BP is similar to EBA with subepidermal blistering and a linear deposit of IgG and C3 at the BMZ. Indirect IF is most commonly used on salt-split-skin to differentiate the two conditions, where autoantibodies are detected on the dermal side of the split in EBA and on the epidermal or both sides in BP. If differentiation is still unclear, Western immunoblot testing, immunoelectron microscopy (IEM) or type VII collagen ELISA (enzyme linked immunoassay studies) can be performed [7]. BP tends to be more responsive to corticosteroid treatment whereas EBA tends to be resistant. EBA has been shown to respond to Dapsone, Cyclosporin A, Mycophenylate, photophoresis and Rituximab [8].
Related posts:
 An Elderly Patient with a Generalized Pruritic Eruption
A Healthy African Child with Blisters
An Elderly Patient with a Generalized Pruritic Eruption
A Healthy African Child with Blisters
 A 52 Year Old Man with Cerebriform Vegetating Masses on the Scalp
A 52 Year Old Man with Cerebriform Vegetating Masses on the Scalp
 Single Step Multivariant Analysis of Serum Autoantibodies in Autoimmune Blistering Diseases Using BIOCHIP® Mosaic Technology
Single Step Multivariant Analysis of Serum Autoantibodies in Autoimmune Blistering Diseases Using BIOCHIP® Mosaic Technology
 A Chronically Ill Teenager with Blisters and Scars
A Chronically Ill Teenager with Blisters and Scars
 Life-Threatening Pemphigus Vulgaris
Life-Threatening Pemphigus Vulgaris
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


