div class=”ChapterContextInformation”>
4. A 6 Years Old Male with Multiple Black Spots on Face
Keywords
Childhood lentiginosisFamilial lentiginosis syndromeMucocutaneous pigmentationSerine/threonine kinase II gene mutationGastrointestinal polypMalignancies- 1.
LEOPARD syndrome
- 2.
Peutz-Jegher’s syndrome
- 3.
Addison’s disease
- 4.
Laughier-Hunziker syndrome
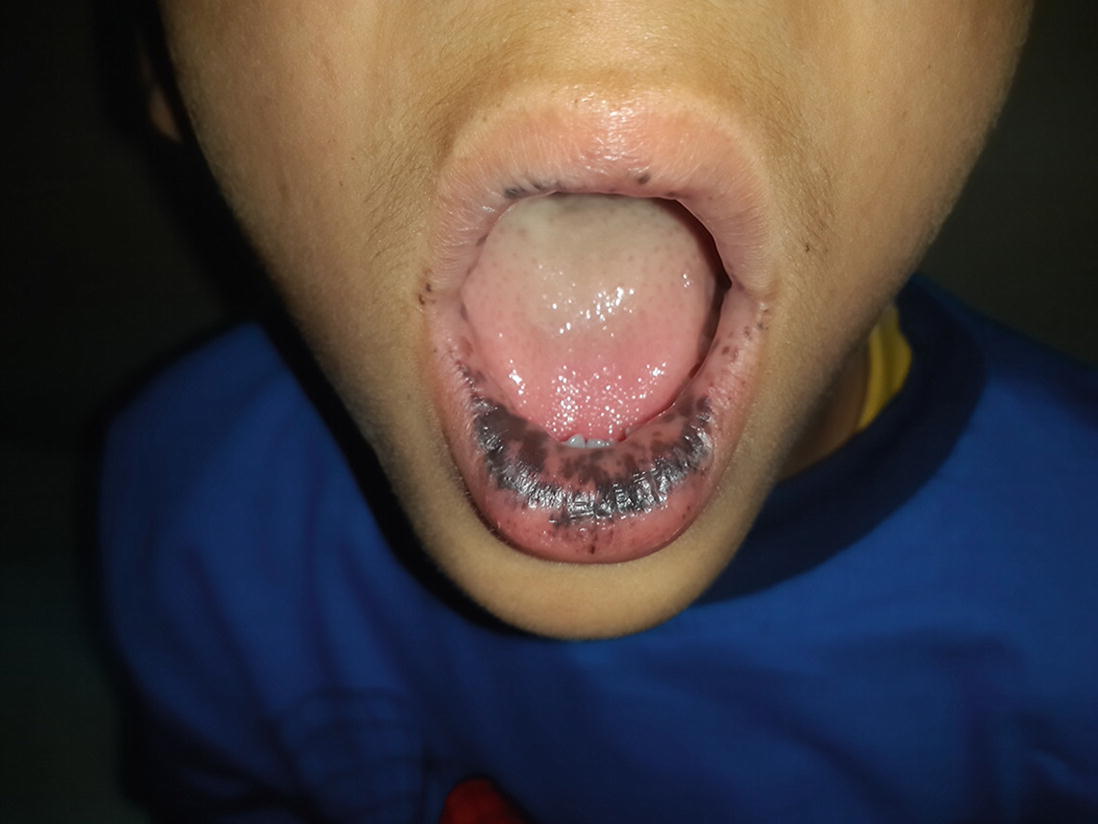
Young boy having dark brown coloured discrete and coalescing macule over lips. (Courtesy: Dr. Kanyarani Vashishth)
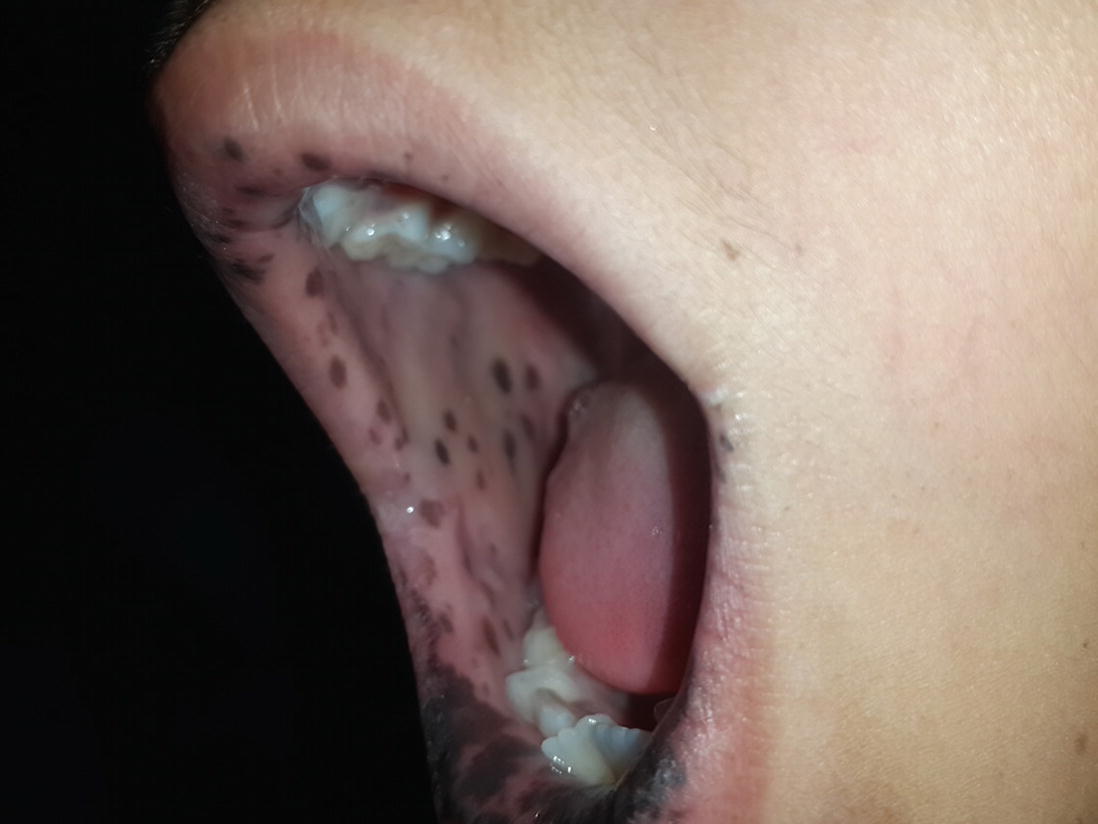
Pigmented macules on buccal mucosa. (Courtesy: Dr. Kanyarani Vashishth)
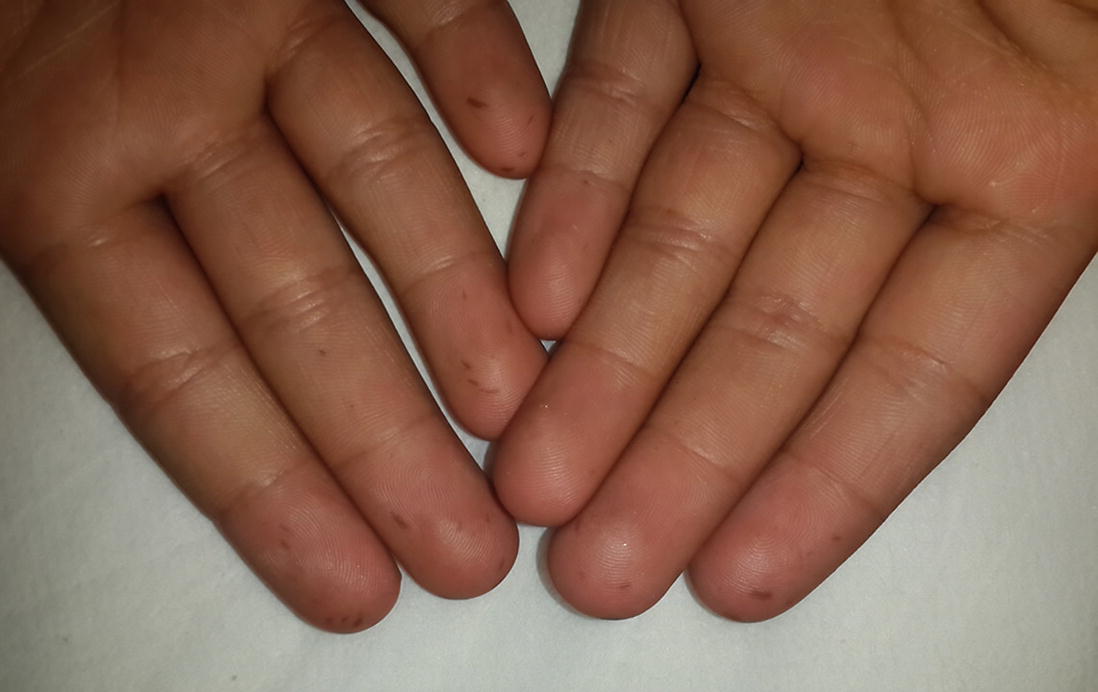
Pigmented macules of Peutz-Jegher’s syndrome on palms. (Courtesy: Dr. Kanyarani Vashishth)
Diagnosis
Peutz-Jegher’s syndrome
Discussion
Peutz-Jegher’s syndrome (PJS), also known as periorificial lentiginosis is a familial lentiginosis syndrome with autosomal dominant inheritance without any sex and racial preponderance. The condition develops due to mutation of a serine/threonine kinase 11gene (LKB1/STK11), located on chromosome 19p13 [1] and is characterised by mucocutaneous pigmentation, gastrointestinal polyps and increased risk of malignancies.
At birth or infancy onwards, dark brown-blue to brown-black pigmented macules of 2–4 mm size develop over lips, gums, buccal mucosa, hard palate, perioral, perinasal, periorbital, perianal skin, palms, soles, dorsum of finger and toes. Additionally, nails may show melanonychia. Except for mucosal lesions, these pigmented macules tend to fade away with time and by adulthood. Localization of pigmented macules in the oral mucosa is characteristic and very suggestive of Peutz-Jegher’s syndrome. The diagnostic criteria put forward by World Health Organization (WHO) allows diagnosis of PJS if characteristic mucocutaneous pigmentation is present in a person with a family history of PJS.
Another hallmark of the disease is multiple benign polyps of the gastrointestinal tract. The common sites of affection are small intestine (64% in order of jejunum, ileum and duodenum), colon (63%), stomach (49%) and rectum (32%). The symptoms related to gastrointestinal polyposis are anaemia, vomiting, rectal bleeding, haematemesis, melaena, repeated abdominal colic, obstruction (usually of small intestine) and/or intussusception, infarction and extrusion of polyp. Symptomatic polyps usually present for the first time in adolescence and early adulthood; up to one-third of patients experience polyp-related symptoms by 10 years of age. Gastric outlet obstruction may present as early as in the neonatal period. Abdominal pain due to polyps causing subtotal obstruction increases in frequency and in intensity with age. Affected patients may develop extraintestinal polyps too; common manifestations include nasal polyps, gall bladder polyps, ureteric polyps, and respiratory tract polyps. These extraintestinal polyps too contribute to morbidity in patients with PJS [2].
The most feared complication is increased risk of malignancies both intestinal and extraintestinal. In general, risk of developing malignancy increases with age and is greater in females than in males. The most commonly reported include colon (39%), small intestine (13%), pancreatic (11–36%), stomach (29%), breast (45–50%), ovary (18–21%), and uterus (9%) cancers. Testicular carcinoma, lung carcinoma and thyroid carcinoma also have been reported [3]. Periodic follow up with endoscopy, colonoscopy, barium follow-through, biopsy and pelvic examination is required.
Diagnostic criteria based on the National Comprehensive Cancer Network (NCCN) 2018 guidelines includes presence of two or more of the following features: at least two biopsy proven Peutz-Jegher’s-type hamartomatous polyps of the gastrointestinal (GI) tract; mucocutaneous hyperpigmentation affecting the eyes, nose, mouth/lips, fingers, or genitals; and a family history of Peutz-Jegher’s syndrome [4].
Table 4.1 Summary of differential diagnosis of Peutz-Jegher’s syndrome
Related posts:
 Years Old Male with Multiple Hyperpigmented Macules on Trunk
Years Old Male with Multiple Hyperpigmented Macules on Trunk
 Gray Pigmented Macule on Right Cheek
Gray Pigmented Macule on Right Cheek
 Female with Multiple Pigmented Macules on Face
Female with Multiple Pigmented Macules on Face
 Young Female with Generalized Mottled Pigmentation
Young Female with Generalized Mottled Pigmentation
 Young Boy with Generalized Hyperpigmentation
Young Boy with Generalized Hyperpigmentation
 Female with Freckles Like Pigmentation on Face and Extremities
Female with Freckles Like Pigmentation on Face and Extremities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


