3 Genital Surgery in Children
3.1 Correction of Congenital Urogenital or Anorectal Malformations in Children
Note
The correction of congenital urogenital and/or anorectal malformations in children should only be undertaken in recognized pediatric surgical centers with a high level of expertise. The treatment of congenital urogenital and/or anorectal malformations initially focuses on primary reconstruction and function. However, as the patient grows and develops, the aesthetic genital surgeon can be confronted with a variety of clinical pictures and their late complications. This chapter is only intended to provide an introduction to the many pediatric malformations and their therapy.
3.1.1 Anorectal Malformations
Fundamentals
Anorectal malformations occur in about 1:5,000 live births; boys are affected more often than girls (male: female = approximately 1.3:1). Findings include anal, rectal, or colonic malformations of varying lengths. These malformations can end in a cul-de-sac or in a fistula communicating with the perineum or urogenital tract (▶Fig. 3.1).
The etiology of these malformations remains unclear. In embryologic terms, growth of the urorectal septum against the cloacal membrane is impaired, which normally occurs within weeks 4 to 8 of pregnancy. However, these blastogenetic defects often include two or more areas, giving rise to complex phenotypes of multifocal origin that include chromosomal, monogenetic, and teratogenic causes. 1 , 5
Of the chromosomal anomalies associated with anorectal malformations, the majority are trisomy 13, trisomy 18, trisomy 21, and partial trisomies and monosomies as well as other chromosomal anomalies. Other familiar syndromes include the Cat Eye syndrome, Townes–Brocks syndrome, FG syndrome (also known as Opitz–Kaveggia syndrome), Pallister–Hall syndrome, the VACTERL association (vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal anomalies, and limb abnormalities), sirenomelia, caudal regression syndrome, and the Currarino triad. 1 , 5
Associated Malformations
Associated malformations occur in 50 to 60% of patients with anorectal malformations. Usually, they involve the urogenital tract. Malformations are significantly more common in boys (26%) than in girls (only 5%). Malformations of the upper urinary tract occur in 50% of the boys and 30% of the girls. The prevalence of these simultaneous urogenital malformations depends on the location of the anorectal malformation. The rate with cloacal malformations is 90%, whereas mild forms of anorectal malformation with a perineal fistula occur in only 10%. These associated urogenital malformations can include the entire spectrum of malformations of the urogenital tract. The most common are vesicoureteral reflux, unilateral renal agenesis, megaureter, and hypospadias.
Associated skeletal malformations usually involve the sacrum and spine. The combination of anorectal malformation, sacral malformation, and the presence of a presacral mass (such as anterior meningocele, teratoma, dermoid cyst, or rectal duplication) is referred to as the Currarino triad. It is associated with a poor functional prognosis for young patients. Less common findings include gastrointestinal malformations, found in 10 to 25% of all cases, or cardiac malformations.
The simultaneous occurrence of at least two of the associated malformations is referred to as the VATER or VACTERL anomaly.
Classification
The broad variation of anorectal malformations has led to proliferation of classification systems. The most common classification system is the Krickenbeck classification according to the anatomic position of the fistula (see ▶Table 3.1). The common malformations are found in the main group; the secondary group includes less common malformations with their regional differences.
Main group boys | Main group girls |
Perineal fistula | Perineal fistula |
Rectourethral fistula
| Vestibular fistula |
Rectovesical fistula | Cloaca |
Anorectal malformation without fistula | Anorectal malformation without fistula |
Anal stenosis | Anal stenosis |
Rare malformations
| |
The Krickenbeck classification (▶Table 3.1) can also be used to determine therapeutic strategies and prognoses. 8
Male Malformations (▶Fig. 3.2a-d)
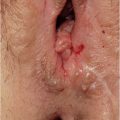
▶Perineal fistula. The lowest part of the rectum lies anterior to the middle of the sphincter, and the opening of the anus is displaced anteriorly. It is usually narrow and may be covered by a membrane. The sphincter and sacrum are normal (▶Fig. 3.2b).
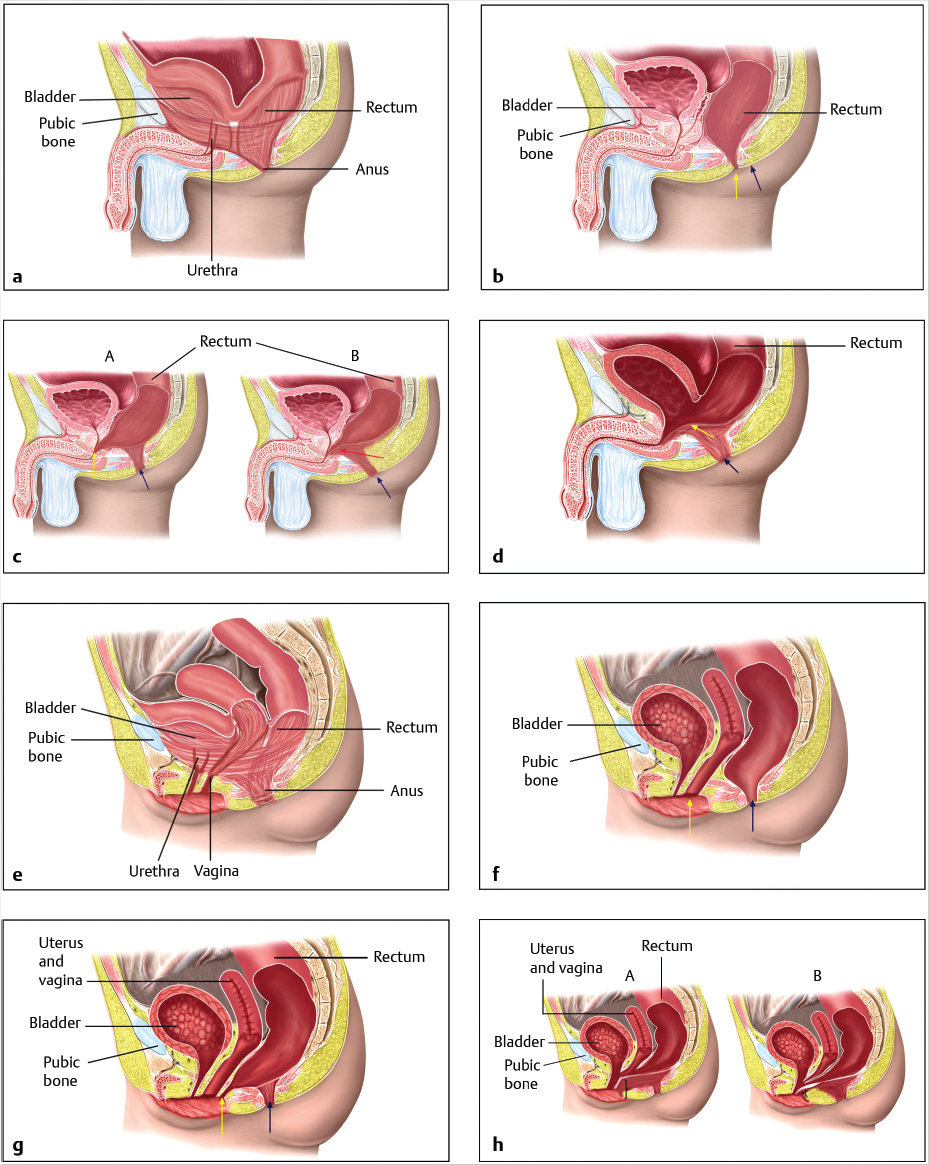
▶Rectourethral fistula. The rectum opens into the posterior urethra, usually the bulbar urethra (low form; ▶Fig. 3.2c A) or, less often, the prostatic urethra (= high form; ▶Fig. 3.2c B). The rectum and urethra have a common wall proximal to the fistula. The higher the fistula is, the shorter the common wall will be. In the low form, the rectum is partially surrounded by the levator ani musculature. The sphincter lies beneath the rectum. The rectobulbar fistula is associated with well-developed musculature, a normal sacrum, and a clearly visible anal fossa. The high form exhibits poorly developed musculature, an abnormal sacrum, a flat perineum, and no anal fossa.
▶Rectovesical fistula. The rectum opens into the neck of the bladder. The sphincter muscle is poorly developed, the perineum is flat, the sacrum is hypoplastic, and the pelvis is underdeveloped (▶Fig. 3.2d).
▶Anorectal malformation without a fistula. The rectum terminates in a cul-de-sac about 3 mm proximal to the perineal skin. Good sphincter musculature is present.
Female Forms (▶Fig. 3.2e–h)
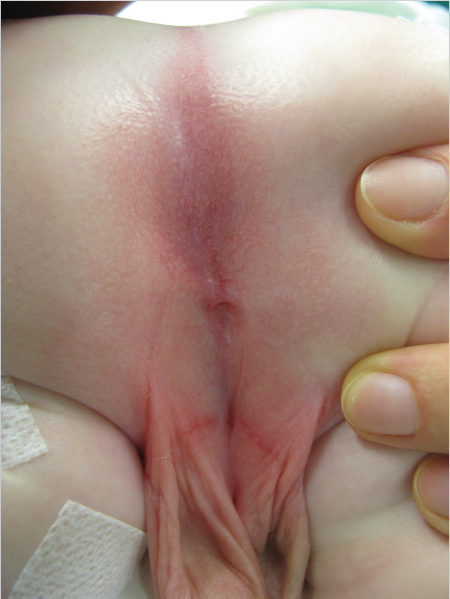
▶Perianal fistula. The opening of the rectum is anteriorly displaced with a narrow fistula between the external female genitals and the center of the sphincter. The sphincter and sacrum are normal (▶Fig. 3.2f).
▶Vestibular fistula. The rectum opens into the vestibule of the vagina directly behind the hymen. The length of the fistula can range from a few millimeters to several centimeters. The rectum and vagina have a common wall proximal to the fistula. The sphincter musculature is usually very well developed and the sacrum is normal (▶Fig. 3.2g).
▶Cloacal malformation. The incidence is about 1 in 50,000 births. This condition is defined as a severe malformation lacking separation of the rectum, vagina, and urethra, and resulting in a common channel. The length of the common channel varies between 1 and 10 cm and correlates with the severity of the malformation and the functional prognosis (▶Fig. 3.2h). 15 In 1987, Pena classified the six most common forms of cloacal malformation with varying length of the common channel and varying configuration of the vagina (or vaginas) and the rectum. The rule of thumb today is that a common channel over 3.5 cm in length is a sign of a complex defect.
▶Anorectal malformation without a fistula. The rectum terminates, as in boys, in a cul-de-sac about 3 mm proximal to the perineal skin. Here, too, good sphincter musculature is present.
Diagnostic Workup
Anal atresia is usually detected immediately after birth during the examination of the newborn.
Note
A surgical treatment strategy must be available within 48 hours.
After the physical examination with inspection of the perineum, an abdominal ultrasound examination is performed to evaluate the urinary tract. Echocardiography is then performed to exclude a cardiac malformation, and the continuity of the esophagus is verified. The newborn initially receives parenteral nutrition and is observed for 24 to 48 hours to determine whether stool passed through the fistula or is in the urine.
A perianal fistula can be carefully dilated to improve stool passage, depending on the nature of stool passage and the width of the fistula. A colon contrast enema will provide information about a possible bowel dilation. If stool passage is uncomplicated and there is no dilation of the bowel proximal to the fistula, then a posterior sagittal anorectoplasty according to Pena and deVries 12 can be planned for the age of one month. However, if bowel dilation is present and stool passage is difficult, then an initial colostomy should be performed immediately. The posterior sagittal anorectoplasty is then performed at the age of 1 to 2 months, after first obtaining new contrast studies visualizing the distal bowel tract with the fistula. If meconium is passed through the urethra or female genital tract, then a colostomy is also performed immediately and a posterior sagittal anorectoplasty at the age of 1 to 2 months.
If there is no fistula or if the findings are equivocal, then a lateral radiograph of the rectum obtained with a horizontal beam with the patient positioned prone and the pelvis elevated should be obtained after 24 to 48 hours. If the lowest point of the rectum is visible distal to the coccyx, or if the distance between the rectum and the skin is less than 1 cm, then a primary posterior sagittal anorectoplasty can be performed immediately. If the rectum is proximal to the coccyx, or if the distance between the rectum and the skin is greater than 1 cm, then an initial colostomy should be performed. In this case, the posterior sagittal anorectoplasty should be carried out as a secondary procedure. Where a cloaca is present, a micturating cystourethrogram and a genitogram should be obtained prior to performing the colostomy. At the age of 3 months, reconstruction with posterior sagittal anorectoplasty, vaginoplasty, and urethroplasty is performed.
Note
High and complicated forms of anorectal malformation should always be corrected only after performing a protective colostomy.
Treatment
Colostomy
The ideal colostomy in anorectal malformation is located at the junction of the descending colon with the sigmoid. The two stomas should be separated by a bridge of skin to prevent stool from entering the distal bowel. Otherwise, it could cause a recurrent urinary tract infection where a simultaneous rectourethral fistula is present, or a megarectum could develop. The bridge of skin also makes it easier to care for the colostomy. If the colostomy is created too far proximally, there will be insufficient bowel compression. This leads to dilation of the distal bowel with irreversible hypomotility and a poor long-term result.
Posterior Sagittal Anorectoplasty According to Pena and deVries
Today, anorectal malformation is usually corrected by performing a posterior sagittal anorectoplasty. This procedure was first described in 1982 by Pena and deVries. This technique allows the surgeon to expose all anatomic components and to correctly pull the rectum through the existing musculature in the center in order to achieve an optimal functional result. 1 , 8
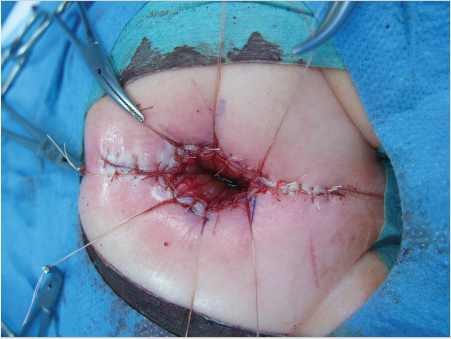
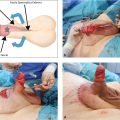
Important steps include positioning the child prone, placing a urinary catheter, electrically stimulating the muscle complex, performing a longitudinal or dragon-shaped skin incision above the anal fossa or the fistula, and extending the incision to the coccyx if necessary. The superficial portion of the sphincter is medially transected, and the fistula is mobilized. Then the rectum is dissected, taking care to spare the urethra. Once a sufficient length of rectum has been obtained (▶Fig. 3.3), the musculature is reconstructed. The rectum is sutured to the sphincter. Then an anoplasty is performed, and the rectum is sutured into the skin (▶Fig. 3.4).
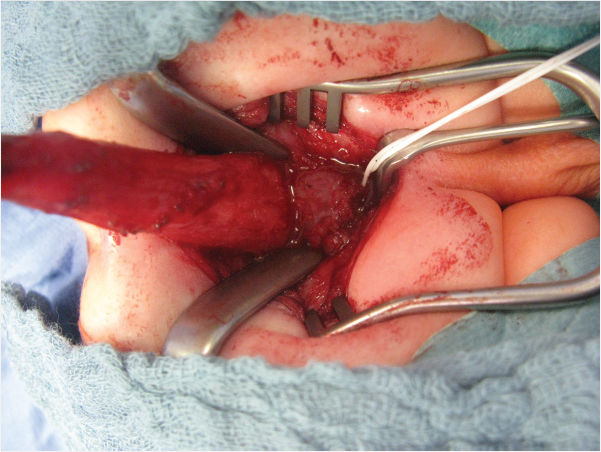
If there is no external fistula, then the dissection begins posteriorly. The parasagittal fibers of the muscle complex are divided, the rectal cul-de-sac is opened at its lowest point, and the fistula is exposed through the rectal lumen. Then the fistula is exposed, mobilized, and ligated and the rectum is further mobilized. In a rectobulbar fistula is a long common wall between the rectum and the urethra which must be split into two layers. The length of this common wall is shorter in the rectoprostatic form, thus, care must be taken to avoid denervation of the bladder or harming the ductus deferens or the seminal vesicles. The bladder neck fistula should be exposed and ligated laparoscopically. In girls, the correction of the perianal fistula or anorectal malformation is performed in the same manner. The retrovestibular fistula is also characterized by a long common wall between the rectum and vagina, which must also be divided.
If the cloacal malformation exhibits a common channel longer than 3 cm, then the posterior sagittal reconstruction with anorectoplasty, vaginoplasty, and urethroplasty is performed similarly to the posterior sagittal anorectoplasty (see above). Also in this case the rectum is exposed posteriorly and then opened posteriorly to expose the common channel. After placement of a transurethral catheter, the vagina and rectum are separated. Then all the urogenital structures are mobilized and moved inferiorly. This is done without separating the urethra and vagina. About 60% of all cloacal malformations can be reconstructed in this manner. However, if the common channel is longer than 3 cm, then the urethra and the vagina must be separated. This requires experience with the various vaginal reconstructive procedures.
Aftercare
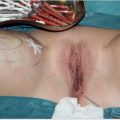
The urinary catheter can be removed 5 days postoperatively or, if the urethra was reconstructed, after 10 days. After 14 days, the neoanus is calibrated and dilation therapy follows immediately (▶Fig. 3.5). The colostomy is usually closed just after 3 months or earlier if sufficient width of the anal canal has been achieved.

Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


