The Ichthyoses: Introduction
|
The ichthyoses are a heterogeneous group of skin diseases characterized by generalized scaling. Scaling reflects altered differentiation of the epidermis. In this chapter, the terms epidermal differentiation, keratinization, and cornification will be used synonymously. The name ichthyosis is derived from the Greek ichthys, meaning “fish,” and refers to the similarity in appearance of the skin to fish scale. Both inherited and acquired forms are found. Early reports of ichthyosis in the Indian and Chinese literature date back to several hundred years bc, and the condition was discussed by Willan in 1808.1
Ichthyosis can present at birth or develop later in life. It can occur as a disease limited to the skin or in association with abnormalities of other organ systems. A number of well-defined types of ichthyosis with characteristic features can be reliably diagnosed. However, because of the great clinical heterogeneity and the profound effect of the environment on scaling, a specific diagnosis can be challenging in certain patients and families.
Siemens introduced genetic concepts into the ichthyoses.2 Wells and Kerr classified the ichthyoses on a genetic basis3 and separated X-linked recessive ichthyosis from ichthyosis vulgaris (IV).4 Gassman developed the concept of retention versus hyperproliferation hyperkeratosis5. Van Scott, Frost, and Weinstein subsequently proposed a classification of the ichthyoses based on differences in rates of epidermal turnover, characterizing them as either disorders of epidermal hyperproliferation or disorders of prolonged retention of the stratum corneum.6 Subsequently, Williams and Elias proposed a classification that lists the disorders of cornification in which clinical, genetic, or biochemical data suggest a distinct disease.7 Traupe reviewed the ichthyoses and suggested classification on a clinical level.5
Genetic approaches to understanding the ichthyoses have revealed the gene defects underlying many of these genodermatoses.8 A new classification is evolving based on the underlying molecular and genetic bases of these disorders. Knowing which gene is mutated directs us to the underlying pathophysiologic process. Understanding and describing these disorders on the basis of common molecular processes leads to more rational approaches to understanding their pathophysiology and treatment. A listing of the more common and the better understood hereditary ichthyoses according to pattern of inheritance and clinical features is shown in Tables 49-1, 49-2, and 49-3. Grouping these disorders according to underlying gene defect (Table 49-4) facilitates understanding of the clinical phenotypes in terms of underlying mechanism. However, further work is necessary to clearly understand how the gene mutations and resultant protein disruptions result in clinical disease, and furthermore, how therapeutic interventions can be creatively developed.
A—Autosomal Dominant/Semidominant | |||||||
|---|---|---|---|---|---|---|---|
Autosomal Semidominant | |||||||
Diagnosis | Onset | Characteristic Clinical Features | Associated Features | Gene | Protein | Function | Skin Histopathology |
Ichthyosis vulgaris OMIM #146700 | Infancy/childhood | Fine or centrally tacked-down scale with superficial fissuring. Relative flexural sparing; worse on lower extremities. Hyperlinear palms/soles. | Keratosis pilaris; atopy | FLG | Absence of filaggrin | Uncertain | Hyperkeratosis; reduced or absent granular layer |
Autosomal Dominant | |||||||
EHK (bullous CIE) OMIM #113800 | Birth | Heterogeneous. May have verrucous, firm, hyperkeratotic (hystrix) spines, often linearly arrayed in flexural creases; blisters; may have erythroderma and/or palmar/plantar keratoderma | Frequent skin infections; characteristic pungent odor | KRT1, KRT10; in Vörner type (confined to palms/soles) KRT 9 | Keratin 1 or 10; in Vörner type, keratin 9 | Structural protein abnormality leading to keratin intermediate filament dysfunction—epidermal fragility | Hyperkeratosis; vacuolated degeneration of the epidermal granular (and often deeper) layer; large, irregular keratohyalin granules |
Ichthyosis bullosa of Siemens OMIM #146800 | Birth | Redness and blistering at birth. Later develop hyperkeratosis, accentuated over flexures. Mauserung (molting): collarette-like lesion where uppermost epidermis has been lost. | KRT2 | Keratin 2, which is expressed in superficial epidermis | As above for EHK | Similar to epidermolytic hyperkeratosis, but confined to the upper spinous and granular layer | |
Ichthyosis hystrix of Curth and Macklin OMIM #146590 | Birth | Resembles EHK, varies from palmar/plantar keratoderma to generalized. Thick “porcupine-like” spines. No blistering. | KRT1—mutation in variable region | Keratin 1 | As above for EHK | Upper epidermal keratinocytes have perinuclear vacuolization/perinuclear shells of tonofilaments | |
Annular epidermolytic erythema OMIM #607602 | Birth/infancy Annular plaques develop later | Severe, intermittent scaling with blisters or pustules. Evolves to widespread, migratory, annular plaques. | KRT1, KRT10 | Keratin 1 or 10 | As above for EHK | As above for EHK | |
Erythrokeratodermia variabilis— OMIM #133200 | |||||||
Generalized type | Birth | Generalized hyperkeratosis and figurate, migratory red patches | Red patches move over minutes to hours, may be triggered by changes in temperature | GJB3 or GJB4 | Connexin 31 or 30.3; connexins form gap junction channels between cells | Abnormal intercellular communication | Hyperkeratosis, acanthosis, papillomatosis, capillary dilatation; epidermis may have “church spire” appearance |
Localized type | Variable | Localized hyperkeratotic plaques with figurate, migratory red patches. | Hyperkeratotic plaques may be induced by trauma. Considerable intrafamilial variability | ||||
Progressive symmetric erythrokeratodermia OMIM #602036 | Shortly after birth | Erythematous, scaly plaques, symmetrically distributed over extremities, buttocks, and face; stabilize in early childhood. Trunk tends to be spared. | LOR or GJB4 | Connexin 30.3 | Cornified envelope precursor | Nonspecific | |
Keratitis–ichthyosis–deafness (KID) syndrome Recessive has been reported OMIM #148210 | Birth/infancy | Progressive corneal opacification; either mild generalized hyperkeratosis or discrete erythematous plaques, which may be symmetric; neurosensory deafness. | Follicular hyperkeratosis, scarring alopecia, dystrophic nails, susceptibility to infection | GJB2, GJB6 | Connexin 26, 30; connexins form gap junction channels between cells | Abnormal intercellular communication | Nonspecific |
Diagnosis | Onset | Characteristic Clinical Features | Associated Features | Gene | Protein | Function | Skin Histopathology |
|---|---|---|---|---|---|---|---|
X-linked recessive ichthyosis OMIM #308100 | Birth/infancy | Fine to large scales; comma-shaped corneal opacities on posterior capsule | Cryptorchidism; female carriers may have corneal opacities and delay of onset or progression of labor in affected pregnancies | STS | Steroid sulfatase | Lipid metabolism—abnormal cholesterol metabolism with accumulation of cholesterol sulfate | Hyperkeratosis, may have hypergranulosis; nonspecific |
Chondrodysplasia punctata X-linked recessive (CDPX) OMIM #302950 | Birth | May begin as erythroderma, linear or whorled atrophic areas or hyperkeratosis, alopecia, skeletal abnormalities, short stature | Cataracts, deafness | ARSE | Arylsufatase E | Lipid metabolism—ill defined: failure of hydrolysis of sulfate ester bonds | |
X-linked dominant chondrodysplasia punctata (Conradi–Hünermann–Happle syndrome) (CDPX2) OMIM #302960 | Birth | CIE at birth, clears and is replaced by linear hyperkeratosis, follicular atrophoderma and pigmentary abnormalities, stippled calcifications on radiographs | Occurs almost exclusively in females; hair shaft abnormalities, short stature, cataracts | EBP | Emopamil binding protein (EBP)—also known as 3β-hydroxysteroid-Δ8,7-isomerase | Lipid metabolism—abnormal cholesterol biosynthesis | |
CHILD syndrome X-linked dominant OMIM #308050 | Birth | Congenital hemidysplasia, ichthyosiform erythroderma, limb defects | Almost exclusively in females | NSDHL; EBP reported | NSDHL (3 β-hydroxysteroid dehydrogenase); EBP reported | Lipid metabolism—postsqualene cholesterol biosynthesis |
Diagnosis | Onset | Characteristic Clinical Features | Associated Features | Gene | Protein | Function | Skin Histopathology |
|---|---|---|---|---|---|---|---|
ARCI (See Table 49-5) | |||||||
ARCI/LI | Birth; often collodion presentation | Large, plate-like, brown scale over most of the body; accentuated on lower extremities. Ectropion; eclabium; alopecia. Palmar/plantar involvement varies. | Heat intolerance | See Table 49-5 | See Table 49-5 | See Table 49-5 | Hyperkeratosis, acanthosis, may show parakeratosis. Nonspecific. |
ARCI/CIE | Fine, white scale; generalized erythroderma. Palmar/plantar involvement varies. | ||||||
ARCI/overlap | Varies between “lamellar” and “CIE.” | ||||||
Harlequin ichthyosis OMIM #242500 | Birth | Markedly thickened skin with geometric, deep fissures. Survivors develop severe CIE phenotype. | Restricted respiration, feeding; neonatal sepsis; often leads to neonatal death. Failure to thrive. | ABCA12 | ATP-binding cassette, subfamily A, member 12 | Lipid metabolism—Membrane transport, abnormality of lipid metabolism | Massively thickened, orthokeratotic stratum corneum; variable acanthosis; granular layer variably decreased |
Ichthyosis prematurity syndrome OMIM #608649 | Birth | Premature delivery of infants with erythrodermic, edematous, caseous scaling skin resembling excessive vernix caseosa, evolves into dry, scaly skin with follicular accentuation with signs of atopy | Respiratory distress, and transient peripheral eosinophilia. The respiratory signs resolve. | FATP4 (SLC27) | Fatty acid transport protein 4 | Lipid metabolism—fatty acid transport | Trilamellar membrane inclusions are seen in the stratum corneum by electron microscopy. |
Netherton syndrome OMIM #256500 | Birth | Ichthyosis linearis circumflexa or similar to congenital ichthyosiform erythroderma; trichorrhexis invaginata | Atopy; high serum levels of IgE; may have aminoaciduria; failure to thrive | SPINK5 | LEKTI (a serine protease inhibitor) | Protein metabolism—may result in premature desmosomal and profilaggrin degradation in stratum corneum. | Nonspecific |
Sjögren–Larsson syndrome OMIM #270200 | Ichthyosis apparent at birth | Generalized fine to coarse hyperkeratosis; spastic diplegia; mental retardation; retinal glistening white dots | Short stature, seizures | FALDH (ALDH10, ALDH3A2) | Fatty aldehyde dehydrogenase | Lipid metabolism—Fatty aldehyde metabolism | Nonspecific |
Refsum disease OMIM #266500 | Ichthyosis develops in adulthood | Progressive neurologic dysfunction; skeletal, cardiac, and renal abnormalities | Retinitis pigmentosa, elevated plasma phytanic acid | Most PAHX; PEX 7 also reported (see RCPD below) | Phytanoyl-CoA hydroxylase (PhyH) | Peroxisome abnormality— Deficiency of phytanic acid catabolism; results in phytanic acid accumulation. | Lipid-containing vacuoles in basal keratinocytes |
Trichothiodystrophy (Tay syndrome; PIBI(D)S, IBI(D)S, BI(D)S) OMIM #278730 OMIM #601675 OMIM #234050 | Some have ichthyosis, which may be apparent at birth; may have collodion presentation | Brittle hair, Photosensitivity, Short stature, Ichthyosis, Intellectual impairment, microcephaly, recurrent infections | Abnormally low sulfur content of hair. | Majority have defect in ERCC2 (XPD). A few have mutations in ERCC3 (XPB), GTF2H5 (TTDA), or TTDN1. | Most XPD. XPB, TTDA, or TTDN1 in a few | Components of transcription factor TFIIH | Tiger tail banding of shafts under polarizing microscopy; hair shaft abnormalities (trichoschisis, trichorrhexis nodosa-like fraying; ribboning) |
Chanarin–Dorfman syndrome (neutral lipid storage disease) OMIM #275630 | Birth; may have collodion membrane | Generalized scaling, resembles CIE; variable extracutaneous involvement: cataracts, decreased hearing, psychomotor delay. | Severe pruritus; neurologic abnormalities; hepatic abnormalities; lipid droplets in circulating leukocytes | CGI-58 | Unknown | Lipid metabolism— Activates adipose-triglyceride lipase for lipolysis of tryglycerides | Lipid droplets in dermal and epidermal cells, and acrosyringia of eccrine ducts |
Neonatal ichthyosis–sclerosing cholangitis syndrome #6073718 | Birth | Neonatal cholestatic jaundice, mild ichthyosis with fine, white scales. | Scarring alopecia of the scalp and eyebrows, enamel dysplasia | CLDN1 | Claudin 1 | Abnormal tight junction | Intracytoplasmic vacuoles in keratinocytes that do not stain with oil red O |
Multiple sulfatase deficiency OMIM #272200 | Birth | Ichthyosis resembling X-linked recessive | Neurological deterioration; skeletal abnormalities; facial dysmorphism | SUMF1 | Cα-formylglycine generating enzyme | Generates catalytic residue in active site of eukaryotic sulfatases | Nonspecific |
Peeling Skin Syndrome | |||||||
Type A (noninflammatory) OMIM #270300 | Birth | Generalized peeling in superficial, thin flakes | Unknown | Unknown | Unknown | Intracellular split within corneocytes in stratum corneum | |
Type A—acral variant OMIM #609796 | Limited to over hands and feet | TGM5 | Transglutaminase 5 | Protein cross-linking | Nonspecific | ||
Type B (inflammatory) | Presents as CIE; evolves to migratory, scaling patches | Pruritus, elevated IgE, aminoaciduria, short stature | Unknown | Unknown | Unknown | Intercellular split between stratum corneum and granular layer; between keratinocytes | |

Online Mendelian Inheritance in Man, OMIM,10 (trademark Johns Hopkins University), a catalog of human genes and genetic disorders, is a useful reference with links to additional resources.
Clinical Presentation
A specific diagnosis in an individual or family with ichthyosis can help to predict prognosis, is important for genetic counseling, and may direct treatment. Several features are useful in distinguishing different forms of ichthyosis. These include the age of onset, presence of collodion membrane at birth, quality of scale, presence/absence of erythroderma, abnormalities in other parts of the skin (e.g., palms and soles, ectropion, eclabium) and adnexal structures (e.g., alopecia, hair follicle, or shaft abnormality), and involvement of other organ systems. In different types of ichthyosis, the appearance of the surface of the skin may vary. Visible scaling may be seen in some patients, with flakes of stratum corneum varying in size from fine to coarse. There may also be thickening of the skin with or without visible scale. The term hyperkeratosis has been used clinically to describe thickened skin, with or without scale, that reflects thickening of the stratum corneum. A family pedigree may clarify the pattern of inheritance. However, many autosomal dominant diseases [e.g., epidermolytic hyperkeratosis (EHK)] have a high frequency of spontaneous mutation, and the lack of a positive family history does not rule out autosomal dominant inheritance. Alternatively, the presence of parental consanguinity may suggest autosomal recessive inheritance. Light microscopic features are usually diagnostic in epidermolytic hyperkeratosis and can be helpful in selected ichthyoses (e.g., Refsum disease, neutral lipid storage disease, acquired ichthyosis of sarcoidosis and mycosis fungoides), but histopathologic examination may not be useful to distinguish other ichthyoses. In many cases, the clinical diagnosis may be clarified by genetic analysis, although mutations are not always demonstrable. The development of ichthyosis in adulthood may be a marker of systemic disease.
Etiology and Pathogenesis
The fully differentiated end product of the epidermis is the stratum corneum, which is composed of terminally differentiated keratinocytes, corneocytes (“bricks”), surrounded by an intercellular matrix (“mortar”) (see Chapter 47). The corneocyte bricks are protein-enriched, and the intercellular mortar is composed of hydrophobic, lipid-enriched membrane bilayers.7 The keratin-laden corneocytes are thought to be primarily responsible for the resilience and water retention properties of the stratum corneum, while the matrix forms most of the permeability barrier to water loss. The normal stratum corneum undergoes desquamation in an organized and invisible manner, with individual corneocytes separating from each other and shedding as single cells. Ichthyotic skin has an abnormal quality and quantity of scale, the barrier function of the stratum corneum is compromised, and there may be alterations in the kinetics of epidermal cell proliferation (see Chapter 46). The stratum corneum can be viewed as a compartment, with thickening of the stratum corneum being the result of cells entering the compartment at an increased rate, or leaving (corneocyte desquamation) too slowly, or both.
The process of epidermal differentiation is complex and not completely understood. Defects in many different aspects and steps of this process can lead to a similar end result: abnormal stratum corneum and scale. In some of these disorders, the underlying gene defect has been identified. For example, mutations in the genes that encode the suprabasal epidermal keratins, keratins 1 and 10, cause epidermolytic hyperkeratosis.11 Mutations in the gene encoding transglutaminase-1, an enzyme that catalyzes the cross-linking of proteins and attachment of ceramides during the formation of corneocytes, are found in up to 55% of patients with autosomal recessive congenital ichthyosis.12–15 The observation that key components of this process cause ichthyosiform disorders highlights the importance and complexity of normal keratinocyte differentiation.
Steroid sulfatase controls the hydrolysis of cholesterol sulfate in corneocytes and is thought to be important in the regulation of corneocyte desquamation. Deficiency of steroid sulfatase causes X-linked recessive ichthyosis.16,17 The observation that several drugs that lower serum cholesterol (e.g., nicotinic acid, triparanol) can induce ichthyotic skin changes indicates the importance of lipid homeostasis in normal keratinization.7 Further evidence is the identification of mutations in the genes encoding cholesterol biosynthetic enzymes as a cause of X-linked dominant chondrodysplasia punctata and CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome, and genes encoding other aspects of lipid biosynthesis in the autosomal recessive congenital ichthyoses.18 The identification of mutations in SPINK5 (serine protease inhibitor, Kazal type 5), encoding a serine protease inhibitor, in Netherton syndrome confirms a role for proteolysis and protease inhibitors in normal epidermal differentiation.19 The discoveries of connexin abnormalities as causes for erythrokeratodermia variabilis, KID (keratitis, ichthyosis, and deafness) syndrome, and other disorders involving ectodermal tissues highlight the role of intercellular communication for properly functioning skin.20,21 Mutations in the FLG gene result in reduced or absent filaggrin and decreased moisture binding in the stratum corneum of patients with IV.22 Mutations in FLG may also result in more severe clinical phenotype in other skin disorders.23,24
These examples highlight the complexity of the process of forming a normally functioning stratum corneum and demonstrate that diverse abnormalities can result in similar clinical phenotypes of thickened stratum corneum and scaling. How do diverse processes result in similar phenotype? The answers are unclear, although studies suggest that defects in the barrier may result in inflammation and hyperproliferation.25 Furthermore, our evolving understanding of these mechanisms continues to clarify the multisystem, clinical phenotypes observed in several ichthyosiform disorders.
Ichthyosis Vulgaris
Ichthyosis vulgaris (OMIM #146700), the most common ichthyosis, is relatively mild. While infants usually have normal skin, the disease often manifests within the first year. The scale of IV is usually most prominent on the extensor surfaces of the extremities, with flexural sparing (Fig. 49-1). The diaper area tends to be spared. There may be fine, white scales over large areas. Particularly on the lower extremities, which are often the most severely involved area, the scales may be centrally attached, with “cracking” (superficial fissuring through the stratum corneum) at the edges. This turning up at the edges can lead to the skin feeling rough.
Figure 49-1

Ichthyosis vulgaris. Full presentation of IV in individuals with mutations in both alleles of the profilaggrin gene (A, C, E); milder presentation in an individual with a profilaggrin mutation in a single allele (B, D, F). A and B. Fine, white scale on lower abdomen. C–F. Hyperlinear palms. (From Smith FJ et al: Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 38:337, 2006, with permission.)
A number of other findings are commonly observed in association with IV.26 Hyperlinear palms are usually present, and some patients may have palmar/plantar thickening approaching a keratoderma. Keratosis pilaris is common, even in individuals with mild IV, and usually involves the outside of the arms, extensor thighs, and buttocks. Atopy is also frequently observed and can manifest as hay fever, eczema, or asthma. These findings can confound an accurate diagnosis, because hyperlinear palms and keratosis pilaris may be seen in atopic individuals who do not have IV. Rarely, individuals with IV may have hypohidrosis with heat intolerance. There is great variation in the severity of clinical manifestations between affected individuals in the same family. The condition usually worsens in climates that are dry and cold and improves in warm, humid environments where the disease may clear dramatically.
The disease was thought to be autosomal dominant and in a study of English school children was found to affect 1 in 250.4 Recently, mutations in the gene encoding profilaggrin have been found to cause IV. Profilaggrin is the precursor protein for multiple copies of filaggrin, which functions during cornification. The inheritance pattern has been clarified to be semidominant; individuals who carry one mutated allele have a mild phenotype, while those with mutations in both profilaggrin alleles (homozygotes or compound heterozygotes for the mutations) (Fig. 49-1) manifest a severe clinical phenotype. In the Anglo-European population, the prevalence of clinical disease is as high as 1 in 80.22,27,28
It is difficult to distinguish some patients with mild IV from simple dry skin (xerosis). Evolving understanding of this very common condition is beginning to clarify how a spectrum of underlying mutations can cause the diverse clinical severity of dry skin from xerosis to severe IV. In addition, on the basis of skin findings alone, males with severe IV may be difficult to differentiate from those affected with X-linked recessive ichthyosis.26,29 The histopathologic findings of IV (Fig. 49-2A) may, as the clinical severity, be more pronounced, with hyperkeratosis and absent granular layer in individuals with two abnormal alleles.
Figure 49-2

Histopathology. A. Ichthyosis vulgaris. Note absent granular layer. B. X-linked recessive ichthyosis. Note compact hyperkeratosis and accentuated granular layer. (Used with permission from Rob McFarlane, MD). C. Autosomal recessive congenital ichthyosis (CIE). Thickened stratum corneum composed of compact hyperkeratosis with foci of parakeratosis. D. Epidermolytic hyperkeratosis. The stratum corneum is thickened (hyperkeratosis) and there is prominent vacuolar degeneration of suprabasalar epidermis most marked at the granular layer.
Filaggrin is an epidermal protein involved in the aggregation of keratin intermediate filaments30 and retention of moisture in the stratum corneum.31 Keratin filaments form a network, or cell matrix, that gives structural integrity to the epidermal keratinocytes. As keratinocytes mature into corneocytes, the keratin filaments collapse and are cross-linked to the cornified cell envelope. Filaggrin is synthesized as a high-molecular weight precursor, profilaggrin, that contains multiple filaggrin molecules and is localized to keratohyalin granules. Biochemical studies of epidermis from patients with IV have shown absence of or decrease in filaggrin and its precursor, profilaggrin.32,33 In the Anglo-European population, null mutations in the gene encoding profilaggrin (FLG) are very strong predisposing factors for atopic dermatitis. A strong association is also found with individuals who have sensitivity to common allergens, allergic rhinitis, early onset and persistent eczema, and asthma in the presence of atopic dermatitis.27,34
X-Linked Recessive Ichthyosis
In the 1960s, X-linked recessive ichthyosis (OMIM #308100) was distinguished clinically from other ichthyoses.4 Shortly thereafter, the syndrome of placental steroid sulfatase deficiency was described in pregnancies with failure to initiate labor in association with low maternal urinary estrogens. Because the majority of maternal urinary estrogens are derived from the fetal adrenal and are metabolized by the placenta, low levels can reflect fetal abnormalities or death. However, in this condition low levels do not indicate severe fetal morbidity. The association between failure to initiate or progress labor and ichthyosis in the male offspring was not appreciated until 1978.16,17 Steroid sulfatase hydrolyzes sulfate esters, which include cholesterol sulfate and sulfated steroid hormones.35 Sulfated fetal adrenal hormones undergo desulfation to estrogens, which are excreted in maternal urine. The absence of steroid sulfatase enzyme in the fetal placenta leads to low maternal urinary estrogens, and in some pregnancies, to a failure of labor to initiate or to progress normally. In males with X-linked recessive ichthyosis, steroid sulfatase enzyme activity is decreased or absent in many tissues, including epidermis, stratum corneum, and leukocytes, and in cultured fibroblasts.36 In addition, cholesterol sulfate, an enzyme substrate, accumulates in serum and in scale. Carrier females have been found to have leukocyte steroid sulfatase levels intermediate between those observed in normal individuals and those in affected males.
X-linked recessive ichthyosis occurs in approximately 1 in 2,000 to 6,000 males.4,37 Scaling may begin in the newborn period and is usually most prominent on the extensor surfaces, although there is significant involvement of the flexural areas. While the extent and degree of scaling are variable, X-linked ichthyosis can usually be distinguished from IV on clinical criteria.4,38 The latter tends to be associated with hyperlinear palms and soles, keratosis pilaris, and a family history of atopy. X-linked ichthyosis tends to have more severe involvement with larger scale, and comma-shaped, corneal opacities may be present in half of adult patients39,40 (Fig. 49-3). Corneal opacities do not affect vision and may be present in female carriers.40 Affected males have an increased risk of cryptorchidism, and independently they are at increased risk for the development of testicular cancer.41 Histopathologic examination shows compact orthohyperkeratosis, acanthosis, and papillomatosis. The granular layer is usually thickened (Fig. 49-2).

Cholesterol sulfate levels are elevated in the serum, epidermis, and scale,42 and there is increased mobility of β-lipoproteins (low-density lipoproteins) on electrophoresis, a feature that can suggest the diagnosis. Steroid sulfatase is one of a group of arylsulfatases located on chromosome Xp22. More than 90% of the mutations in X-linked ichthyosis are deletions that can often be detected by fluorescence in situ hybridization (FISH), available in many clinical laboratories. Confirmation of the diagnosis can also be made by finding an elevation in serum cholesterol sulfate levels. Unfortunately, cholesterol sulfate determination in serum and scale may not be readily available for laboratory confirmation of the clinical diagnosis. In the rare case where deletions are not detected and a clinical diagnosis is necessary, arylsulfatase C (steroid sulfatase) enzyme activity can be measured in cultured fibroblasts. Deletions that include adjacent sulfatases explain the overlap syndromes involving chondrodysplasia punctata and X-linked ichthyosis.43 The X-linked form of Kallmann syndrome, in which hypogonadotropic hypogonadism and anosmia are found, often with renal abnormalities, obesity, synkinesis (mirror image movements of the extremities), cleft palate, and spastic paraplegia, can also be seen in association with X-linked recessive ichthyosis as part of a contiguous gene deletion syndrome.43 Because of this, and because of the association with testicular carcinoma, patients with X-linked recessive ichthyosis should be queried about anosmia and have periodic testicular examination.44
In the epidermis, steroid sulfatase catalyzes the hydrolysis of cholesterol sulfate. The identification of steroid sulfatase deficiency in X-linked ichthyosis supports the importance of cholesterol sulfate hydrolysis in normal desquamation. Topical application of cholesterol sulfate in mice can induce a scaling disorder, further supporting the role of cholesterol sulfate hydrolysis in corneocyte desquamation.
A family was described with an ichthyosis inherited in an X-linked pattern but with normal steroid sulfatase activity and the absence of corneal opacities.45 This demonstrates heterogeneity within X-linked ichthyosis. Because of its frequent occurrence, steroid sulfatase deficiency accounts for most cases of X-linked ichthyosis, but a normal steroid sulfatase level in a male with ichthyosis does not rule out an X-linked pattern of inheritance.
Autosomal Recessive Congenital Ichthyosis
The term autosomal recessive congenital ichthyosis (ARCI) is useful to describe a heterogeneous group of disorders that present at birth with generalized involvement of the skin. Autosomal recessive ichthyosis is rare and has been estimated to occur in about 1 in 300,000 persons.4
In older literature, nonbullous congenital ichthyosiform erythroderma (NCIE) [lamellar ichthyosis (LI), with autosomal recessive inheritance] was distinguished from bullous congenital ichthyosiform erythroderma (BCIE) (EHK, with autosomal dominant inheritance) based on clinical appearance (bullae) and pattern of inheritance.6,46 That the term LI was used interchangeably with NCIE and included a spectrum of phenotypes has led to some confusion. Williams and Elias distinguished LI from NCIE [usually called congenital ichthyosiform erythroderma (CIE)], a milder erythrodermic form.47 Some patients with LI can be clearly distinguished from those with CIE on the basis of clinical features. In LI one sees large, dark, plate-like scales, and while infants may be red at birth, adults have little to no erythroderma (Fig. 49-4). In the more severe, classic presentation of LI, tautness of the facial skin leads to traction on the eyelids and lips, resulting in ectropion and eclabium. Scarring alopecia, most prominent at the periphery of the scalp, may be partly due to traction at the hairline. In contrast, CIE has generalized redness and fine, white scales (Fig. 49-5). Patients with classic CIE have little to no ectropion, eclabium, or alopecia. However, many patients do not fit neatly into these two clinical descriptions,48 in that they have features of both LI and CIE with a clinical phenotype intermediate between both disorders. Therefore, it can be useful to consider these two distinctive presentations as ends of a spectrum, between which lie a gradation of clinical phenotypes with variable degrees of erythema and coarseness of scale. Individual features such as collodion membrane (discussed below), ectropion, and alopecia can occur across the spectrum. While attempts to refine the categorization of these disorders by biochemical and ultrastructural observations have failed to yield a consistent and replicable classification scheme,5 identification of the spectrum of specific molecular defects underlying these conditions will undoubtedly help.49 Identification of mutations has, so far, found ARCI to be caused by six different genes (Table 49-5) which are important for the formation of the intercellular lipid layer or the cornified envelope of keratinocytes.


Genea | Protein | Function |
|---|---|---|
TGM1b | Transglutaminase 1 | Cornified envelope formation |
ABCA12 | ATP-binding cassette, subfamily A, member 12 | Membrane transport/lipid metabolism |
ALOXE3 | Arachidonate lipoxygenase 3 (eLOX3) | Hydroperoxide isomerase |
ALOX12B | Arachidonate 12-lipoxygenase, R type (12R-LOX) | Lipoxygenase |
ICHYN | Ichthyin | Trioxilin A3 receptor? Hepoxylin pathway? |
FLJ39501 (CYP4F22) | Cytochrome P450, family 4, subfamily F, polypeptide 2 | LTB4-ω-hydoxylase? |
? (12p11.2-q13) | ||
? (19p13.1-13.2) |
Most patients with LI or CIE inherit the disease in an autosomal recessive pattern. Rarely, families have been described with similar phenotypes, where the disease is inherited as an autosomal dominant trait.5 This is an important consideration for genetic counseling.
Lamellar Ichthyosis
LI is apparent at birth, and the newborn usually presents encased in a collodion membrane. At this time the skin may be red. Over time, the skin develops large, plate-like scales, which appear to be arranged in a mosaic pattern (see Fig. 49-4). In some areas, the scales are centrally attached with raised borders. The scales tend to be largest over the lower extremities, where the large, plate-like scales separated by superficial fissuring can lead to an appearance similar to that of a dry riverbed. During childhood and into adulthood the degree of erythema may vary, but usually is minimal or absent in the severe presentation of classic LI. Involvement of the palms and soles in LI is variable and ranges from minimal hyperlinearity to severe keratoderma.
The lips and mucous membranes tend to be spared in LI, but the adnexal structures may be compromised by the adherent, firm scales. Thick stratum corneum on the scalp tends to encase hairs, and in conjunction with the tautness of the skin, may lead to a scarring alopecia, most marked at the periphery of the scalp. The hyperkeratosis can interfere with normal sweat gland function, resulting in hypohidrosis, but the degree of impairment varies between patients. Some patients have severe heat intolerance and must be vigilant to avoid overheating. Treatment with oral retinoids can improve or prevent some sequelae of LI. Patients frequently notice an increase in sweating, with improved heat tolerance. While retinoid therapy can cause blepharitis or even conjunctivitis, it is usually well tolerated by patients with LI. Moreover, the ability of systemic retinoid (and in some cases, topical retinoid) therapy to decrease thick periocular scale can decrease the tendency to develop ectropion. Nevertheless, patients with severe, classic LI usually require careful eye maintenance. Because of the ectropion (see Fig. 49-4), the lids may fail to close fully, particularly during sleep; hydration with liquid tears during the day and ophthalmic lubricants at night can prevent exposure keratitis.
Histopathologic examination typically shows orthokeratotic hyperkeratosis with mild to moderate acanthosis. Rates of epidermal proliferation are normal or only slightly elevated in patients with LI, in contrast to those with CIE, which has significantly greater labeling indices.50
Mutations in TGM1, the gene encoding transglutaminase 1, were found in several families with LI, solidifying the role for transglutaminase 1 in the formation of a normal stratum corneum and its role as a cause of LI.12,13 The lamellar phenotype is more commonly associated with mutations in TGM1.15,49 The transglutaminases catalyze calcium-dependent cross-linking of proteins through the formation of ϵ-(γ-glutamyl)lysine isodipeptide bonds. During the formation of the stratum corneum, transglutaminase catalyzes the cross-linking of cellular proteins, including involucrin, loricrin, small proline-rich proteins, and others. The resulting protein complex is deposited on the inner side of the plasma membrane to form the cornified envelope. Transglutaminase also attaches ceramides secreted into the intercellular space by lamellar bodies to cornified envelope proteins, notably involucrin, and thereby is important in the formation of both the protein and lipid components of the stratum corneum.14
Bathing suit ichthyosis is an unusual form of LI where affected individuals develop the scaling typical of LI, but limited to the bathing suit area.51 The distribution correlates with warmer areas of skin. Decreased transglutaminase is found in these areas, and unique, temperature-sensitive mutations in TGM1 have been identified in affected individuals.52
In a human skin/immunodeficient mouse xenograph model, transfer of a transglutaminase-1 gene into transglutaminase-1-deficient keratinocytes from LI patients resulted in normalization of transglutaminase expression and epidermal architecture in addition to restoration of cutaneous barrier function.53 Additional disease-causing loci have been found (see below).
Congenital Ichthyosiform Erythroderma
As with LI, CIE is apparent at birth, and the newborn usually presents with a taut, shiny, collodion membrane. After shedding of the membrane, the skin of infants with CIE remains red, usually with a fine, white, generalized scale (see Fig. 49-5). On the lower legs, the scale may be larger and darker. In contrast to LI, the classic presentation of CIE may have little to no ectropion, eclabium, or alopecia. As in LI, there is a wide variation in the ability to sweat, and patients with CIE may have minimal sweating with severe heat intolerance. Mucous membranes are usually spared. Palm/sole involvement is variable. Nails may have ridging, but they are often spared. As with all the ichthyoses, dermatophyte infection of the skin and nails is common.
Histopathologic examination shows hyperkeratosis, acanthosis, and often parakeratosis (Fig 49-2C). Studies of epidermal proliferation have shown markedly increased epidermal cell turnover, in contrast to LI.50
A small subset of patients with CIE have been found to have mutations in TGM1, and two siblings with a collodion presentation, CIE, and palmoplantar keratoderma had an autosomal dominant mutation in the cornified envelope protein, loricrin.54 Further genetic heterogeneity within ARCI has been demonstrated, with exclusion of TGM1 as the disease-causing gene in families with phenotypes within the spectrum of classic LI and CIE and identification of mutations in other genes. To date, mutations have been identified in five additional genes in ARCI (Table 49-5). In addition, two other genetic loci (12p11.2-q13 and 19p13.1-13.2) have been found to be associated with ARCI; the responsible genes have not been identified. The enzymes coded for by these genes (Table 49-5) are hypothesized to function together to convert arachidonic acid to a specific hepoxilin in a newly described pathway thought to form a common link in ARCI.18,55 This hypothesis remains to be tested, and how the most common mutation in ARCI, that of TGM1, might relate to lipid abnormalities is unclear.
Epidermolytic Hyperkeratosis
In 1902, Brocq described bullous ichthyotic erythroderma and distinguished the blistering type from the nonblistering type of congenital ichthyotic erythroderma.56 The original description included three unrelated patients whose clinical manifestations varied. However, this was probably the first description of epidermolytic hyperkeratosis (EHK; OMIM #113800). The disease is named for the distinctive histopathologic features of vacuolar degeneration of the epidermis (i.e., epidermal lysis) and associated hyperkeratosis. EHK is also known as BCIE, an earlier descriptive name signifying the blistering, neonatal presentation, scaling, and redness.
EHK is transmitted as an autosomal dominant trait with a prevalence of approximately 1 in 200,000 to 300,000 persons. However, there is a high frequency of spontaneous mutation, and as many as one-half of the cases have no family history46 and represent new mutational events. The disease usually presents at birth with blistering, redness, and peeling (Fig. 49-6). With time, generalized hyperkeratosis may develop, which may or may not be associated with erythroderma. EHK skin usually has a characteristic pungent odor, thought to be related to superinfection by mixed flora.

In contrast to most other ichthyoses, the histopathologic picture of EHK is almost always distinctive. A tremendously thickened stratum corneum and vacuolar degeneration of the upper epidermis are seen, leading to the histologic term EHK (see Fig. 49-2D). The vacuolar degeneration usually involves the upper epidermis and occasionally all of the suprabasilar keratinocytes. Granular cells exhibit dense, enlarged, irregularly shaped masses that appear to be keratohyalin granules.6 On electron-microscopic examination, clumping of filaments is observed to begin in the first suprabasal layer. These aggregated filaments are clumps of keratin intermediate filaments that contain the suprabasal keratins 1 and 10.57
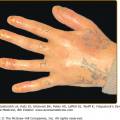
There is striking clinical heterogeneity between EHK families. Six distinctive clinical phenotypes have been described.58 Several clinical features can be useful to distinguish between the different subtypes, including the presence versus absence of severe palmar/plantar hyperkeratosis and of erythroderma, quality of scale, extent of involvement, presence of digital contractures, and gait abnormality (Table 49-6). Three types without palm/sole hyperkeratosis are called NPS types. Patients with the NPS-1 clinical phenotype have generalized involvement with thick, brown verrucous hyperkeratosis, most prominent over joints. Palms and soles are spared. Cobblestone or hystrix (porcupine-like) spiny papules are characteristic; this severe clinical presentation is one of the more commonly recognized types of EHK (Fig. 49-7A). NPS-2 is similar, but much milder, without the hystrix spines and with relative sparing of the skin between joints (Fig. 49-7B). Generalized exfoliative erythroderma is found in NPS-3 (Fig. 49-7C). Three types with severe palm/sole hyperkeratosis are called PS types. Patients with the PS-1 type have a smooth palmar/plantar hyperkeratosis with a sharp border, often delineated by a red halo. Blisters may be present at the border. There may be limited involvement of the trunk, usually confined to areas over joints (Fig. 49-8A). The PS-2 type is characterized by generalized erythroderma and scaling. Volar involvement may be severe, with contractures of the digits and ainhum-like constricting digital bands (Fig. 49-8B). The PS-3 type has generalized skin involvement with a pebbly hyperkeratosis that is arrayed in a distinctive, cerebriform pattern on the palms and soles (Fig. 49-8C).
NPS-1 | NPS-2 | NPS-3 | PS-1 | PS-2 | PS-3 | |
|---|---|---|---|---|---|---|
Palm/sole hyperkeratosis | − | − | − | + | + | + |
Palm/sole surface | Normal | Normal | Hyperlinear, minimal scale | Smooth | Smooth | Cerebriform |
Digital contractures | − | − | − | − | + | − |
Scale | Hystrix | Brown | Fine, white | Mild | White scale to peel | Tan |
Distribution | Generalized | Generalized | Generalized | Localized | Generalized | Generalized |
Erythroderma | − | − | + | − | + | − |
Blistering | + | + | + | Localized | + | Neonatal |
Implied mutation | Keratin 10 | Keratin 10 | Keratin 10 | Keratin 1 | Keratin 1 | Keratin 1 |
Figure 49-7

Clinical phenotypes of epidermolytic hyperkeratosis. NPS (no severe palm/sole hyperkeratosis) types are shown. A. NPS-1 (NPS-type 1): Generalized involvement with thick, brown verrucous hyperkeratosis. Palms are spared. Over the dorsal foot, hyperkeratosis is arrayed in a characteristic cobblestone or hystrix (porcupine-like) pattern. B. NPS-2 (NPS-type 2): Brown hyperkeratosis over the trunk, and accentuated over joints. Palms are spared. Compared to the verrucous hyperkeratosis in NPS-1, involvement is much milder. On the lower legs, note the blistering, hypertrichosis, and relative sparing of the skin between the joints. C. NPS-3 (NPS-type 3): Generalized erythroderma with white scale and hyperkeratosis.
Figure 49-8

PS (Severe palm/sole hyperkeratosis) types are shown. A.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


