Psoriasis: Introduction
|
Epidemiology
 The earliest descriptions of what appears to represent psoriasis are given at the beginning of medicine in the Corpus Hippocraticum. This work was edited in Alexandria 100 years after the death of Hippocrates (460–377 bc), who presumably was the author. Hippocrates used the terms psora and lepra for conditions that can be recognized as psoriasis. Later, Celsus (ca. 25 bc) described a form of impetigo that was interpreted by R. Willan (1757–1812) as being psoriasis. Willan separated two diseases as psoriasiform entities, a discoid lepra Graecorum and a polycyclic confluent psora leprosa, which later was called psoriasis. In 1841, the Viennese dermatologist Ferdinand von Hebra (1816–1880) unequivocally showed that Willan’s lepra Graecorum and psora leprosa were one disease that had caused much confusion because of differences in the size, distribution, growth, and involution of lesions.
The earliest descriptions of what appears to represent psoriasis are given at the beginning of medicine in the Corpus Hippocraticum. This work was edited in Alexandria 100 years after the death of Hippocrates (460–377 bc), who presumably was the author. Hippocrates used the terms psora and lepra for conditions that can be recognized as psoriasis. Later, Celsus (ca. 25 bc) described a form of impetigo that was interpreted by R. Willan (1757–1812) as being psoriasis. Willan separated two diseases as psoriasiform entities, a discoid lepra Graecorum and a polycyclic confluent psora leprosa, which later was called psoriasis. In 1841, the Viennese dermatologist Ferdinand von Hebra (1816–1880) unequivocally showed that Willan’s lepra Graecorum and psora leprosa were one disease that had caused much confusion because of differences in the size, distribution, growth, and involution of lesions.
Psoriasis is universal in occurrence. However, its prevalence in different populations varies from 0.1% to 11.8%, according to published reports.1 The highest reported incidences in Europe have been in Denmark (2.9%) and the Faeroe Islands (2.8%). A recent study of 1.3 million Germans found a prevalence of 2.5%.2 Similar prevalence (ranging from 2.2% to 2.6%) has been measured in the United States. A higher prevalence in East Africans as opposed to West Africans may explain the relatively low prevalence of psoriasis in African-Americans (1.3% vs. 2.5% in white Americans).3 The incidence of psoriasis is also low in Asians (0.4%), and in an examination of 26,000 South American Indians, not a single case was seen. Psoriasis is equally common in males and females.4,5
Psoriasis may begin at any age, but it is uncommon under the age of 10 years. It is most likely to appear between the ages of 15 and 30 years. Possession of certain HLA Class I antigens, particularly HLA-Cw6, is associated with an earlier age of onset and with a positive family history. This finding led Henseler and Christophers6 to propose that two different forms of psoriasis exist: type I psoriasis, with age of onset before 40 years and HLA-associated, and type II, with age of onset after 40 years and lacking HLA associations, although many patients do not fit into this classification. There is no evidence that type I and type II psoriasis respond differently to treatment.
Etiology and Pathogenesis
Psoriasis is a chronic inflammatory skin disease, with a strong genetic basis, characterized by complex alterations in epidermal growth and differentiation and multiple biochemical, immunologic, and vascular abnormalities, and a poorly understood relationship to nervous system function. Its root cause remains unknown. Historically, psoriasis was widely considered to be a primary disorder of keratinocytes. With the discovery that the T-cell specific immunosuppressant cyclosporine A (CsA) was highly active against psoriasis, research became more focused on T cells and the immune system. Nevertheless, accumulating evidence shows that keratinocytes are an integral part of the cutaneous immune reponse in psoriasis.7
 That psoriasis has a genetic basis has been appreciated for nearly 100 years.8 However, as Gunnar Lomholt lamented in 1963 in his classic study of psoriasis in the Faeroe Islands9: “That psoriasis is genetically conditioned is beyond doubt. But when the mode of inheritance appears to have been almost demonstrated, it again slips out of the fetters of fixed rules!” Over the years, based on some very large pedigrees and population surveys, single-gene recessive, two-gene recessive, dominant with reduced penetrance, and polygenic models have been suggested; recurrence risk analysis strongly favors the polygenic model.10 Based on population studies, the risk of psoriasis in an offspring has been estimated to be 41% if both parents are affected, 14% if one parent is affected, and 6% if one sibling is affected, compared to 2% when no parent or sibling is affected.11
That psoriasis has a genetic basis has been appreciated for nearly 100 years.8 However, as Gunnar Lomholt lamented in 1963 in his classic study of psoriasis in the Faeroe Islands9: “That psoriasis is genetically conditioned is beyond doubt. But when the mode of inheritance appears to have been almost demonstrated, it again slips out of the fetters of fixed rules!” Over the years, based on some very large pedigrees and population surveys, single-gene recessive, two-gene recessive, dominant with reduced penetrance, and polygenic models have been suggested; recurrence risk analysis strongly favors the polygenic model.10 Based on population studies, the risk of psoriasis in an offspring has been estimated to be 41% if both parents are affected, 14% if one parent is affected, and 6% if one sibling is affected, compared to 2% when no parent or sibling is affected.11
 The concordance rate for psoriasis in monozygotic twins ranges from 35% to 73%.12–14 This variability, and the fact that these rates do not approach 100%, supports a role for environmental factors. Thus, the mode of inheritance for psoriasis is best described as multifactorial (i.e., polygenic plus environmental factors).15 Interestingly, concordance in both monozygotic and dizygotic twins decreases as one moves closer to the equator.14 Given the beneficial antipsoriatic effects of ultraviolet (UV) light (see Section Treatment), these data suggest that UV exposure may be a major environmental factor interacting with genetic factors in psoriasis.
The concordance rate for psoriasis in monozygotic twins ranges from 35% to 73%.12–14 This variability, and the fact that these rates do not approach 100%, supports a role for environmental factors. Thus, the mode of inheritance for psoriasis is best described as multifactorial (i.e., polygenic plus environmental factors).15 Interestingly, concordance in both monozygotic and dizygotic twins decreases as one moves closer to the equator.14 Given the beneficial antipsoriatic effects of ultraviolet (UV) light (see Section Treatment), these data suggest that UV exposure may be a major environmental factor interacting with genetic factors in psoriasis.
 The search for the involvement of specific genes in psoriasis began two decades ago with studies of genetic linkage (i.e., the cotransmission of marker and disease alleles within families). These studies had the advantage that only a relatively small number of genetic markers (hundreds) were needed to scan the genome. However, linkage studies require collection of large families, which are not that commonly encountered in practice and lack power when disease allele frequencies are high and penetrances are low, as is the case in polygenic disorders16 (see Chapter 8). Despite multiple genome-wide linkage studies, only one locus, termed psoriasis susceptibility 1 (PSORS1), was consistently confirmed in the “linkage era.”17 PSORS1 is located in the major histocompatibility complex (MHC, chromosome 6p21.3), home of the HLA genes. Multiple HLA alleles have been associated with psoriasis, particularly HLA-B13, HLA-B37, HLA-B46, HLAB57, HLA-Cw1, HLA-Cw6, HLA-DR7, and HLA-DQ9.10 Many of these alleles are in linkage disequilibrium with HLA-Cw6 (i.e., found together on the same chromosome more often than would be predicted by chance). Psoriasis is consistently associated with the Class I, rather than the Class II, end of extended HLA haplotypes carrying these risk alleles.18–20 HLA-Cw6 has consistently demonstrated the highest relative risk for psoriasis in Caucasian populations,10 and remains strongly associated with psoriasis when found together with several different HLA-B alleles,21 suggesting that the PSORS1 gene must reside telomeric to HLA-B. HLA-Cw6 is also associated with psoriatic arthritis, with a tendency for early onset of skin lesions.22 HLA-B27, HLA-B38, and HLA-B39 are also associated with psoriatic arthritis even in the absence of HLA-Cw6,23 with HLA-B27 being most strongly associated with the axial variant (see Chapter 19).
The search for the involvement of specific genes in psoriasis began two decades ago with studies of genetic linkage (i.e., the cotransmission of marker and disease alleles within families). These studies had the advantage that only a relatively small number of genetic markers (hundreds) were needed to scan the genome. However, linkage studies require collection of large families, which are not that commonly encountered in practice and lack power when disease allele frequencies are high and penetrances are low, as is the case in polygenic disorders16 (see Chapter 8). Despite multiple genome-wide linkage studies, only one locus, termed psoriasis susceptibility 1 (PSORS1), was consistently confirmed in the “linkage era.”17 PSORS1 is located in the major histocompatibility complex (MHC, chromosome 6p21.3), home of the HLA genes. Multiple HLA alleles have been associated with psoriasis, particularly HLA-B13, HLA-B37, HLA-B46, HLAB57, HLA-Cw1, HLA-Cw6, HLA-DR7, and HLA-DQ9.10 Many of these alleles are in linkage disequilibrium with HLA-Cw6 (i.e., found together on the same chromosome more often than would be predicted by chance). Psoriasis is consistently associated with the Class I, rather than the Class II, end of extended HLA haplotypes carrying these risk alleles.18–20 HLA-Cw6 has consistently demonstrated the highest relative risk for psoriasis in Caucasian populations,10 and remains strongly associated with psoriasis when found together with several different HLA-B alleles,21 suggesting that the PSORS1 gene must reside telomeric to HLA-B. HLA-Cw6 is also associated with psoriatic arthritis, with a tendency for early onset of skin lesions.22 HLA-B27, HLA-B38, and HLA-B39 are also associated with psoriatic arthritis even in the absence of HLA-Cw6,23 with HLA-B27 being most strongly associated with the axial variant (see Chapter 19).
 A haplotype containing HLA-Cw1 and HLA-B46, found only in Southeast Asian populations, is strongly associated with psoriasis in Thailand and Japan.18,24,25 In these populations, approximately one-third of psoriatics are HLA-Cw6-associated, one-third are HLA-Cw1-B46-associated, and one-third show no detectable HLA association.18 Unlike HLA-Cw6, which is associated with early age of onset in Southeast Asian as well as Caucasian psoriatics, the HLA-Cw1-B46 haplotype is equally prevalent in early- and late-onset psoriatics.18 Although HLA-Cw6 is strongly associated with guttate psoriasis,26 there is no evidence for a similar association with the HLA-Cw1-B46 haplotype. Interestingly, this haplotype has been associated with other autoimmune diseases, including myasthenia gravis and Grave’s disease.27
A haplotype containing HLA-Cw1 and HLA-B46, found only in Southeast Asian populations, is strongly associated with psoriasis in Thailand and Japan.18,24,25 In these populations, approximately one-third of psoriatics are HLA-Cw6-associated, one-third are HLA-Cw1-B46-associated, and one-third show no detectable HLA association.18 Unlike HLA-Cw6, which is associated with early age of onset in Southeast Asian as well as Caucasian psoriatics, the HLA-Cw1-B46 haplotype is equally prevalent in early- and late-onset psoriatics.18 Although HLA-Cw6 is strongly associated with guttate psoriasis,26 there is no evidence for a similar association with the HLA-Cw1-B46 haplotype. Interestingly, this haplotype has been associated with other autoimmune diseases, including myasthenia gravis and Grave’s disease.27
 Linkage disequilibrium has posed the major obstacle to confirming or refuting the role of HLA-Cw6 in psoriasis. A particularly strong region of LD containing ten genes is found just telomeric to HLAC,28,29 and not surprisingly, several of these genes also display strong associations with psoriasis. By comparative DNA sequencing of risk versus nonrisk haplotype haplotypes, we were able to exclude all structural variants of all these genes with the exception of HLA-C and CDSN.30 CDSN is an interesting candidate, as its expression is increased in psoriasis, and it plays an important role in regulating the desquamation of keratinocytes.31 By further analysis of genetic markers located between HLA-C and CDSN in Caucasian30 and Chinese32 populations, CDSN could also be excluded, leaving HLA-Cw6 as the only remaining gene candidate for PSORS1. However, additional work is needed to rule out the possibility of regulatory variant(s) in this region that do not fall within genes. It is also likely that additional psoriasis susceptibility genes remain to be identified within the MHC, as haplotypes carrying HLA-Cw6 along with different HLA-B alleles carry different risks of disease.21 Recent studies have mapped one of these additional determinants to the MHC Class II–Class III interface.33,34
Linkage disequilibrium has posed the major obstacle to confirming or refuting the role of HLA-Cw6 in psoriasis. A particularly strong region of LD containing ten genes is found just telomeric to HLAC,28,29 and not surprisingly, several of these genes also display strong associations with psoriasis. By comparative DNA sequencing of risk versus nonrisk haplotype haplotypes, we were able to exclude all structural variants of all these genes with the exception of HLA-C and CDSN.30 CDSN is an interesting candidate, as its expression is increased in psoriasis, and it plays an important role in regulating the desquamation of keratinocytes.31 By further analysis of genetic markers located between HLA-C and CDSN in Caucasian30 and Chinese32 populations, CDSN could also be excluded, leaving HLA-Cw6 as the only remaining gene candidate for PSORS1. However, additional work is needed to rule out the possibility of regulatory variant(s) in this region that do not fall within genes. It is also likely that additional psoriasis susceptibility genes remain to be identified within the MHC, as haplotypes carrying HLA-Cw6 along with different HLA-B alleles carry different risks of disease.21 Recent studies have mapped one of these additional determinants to the MHC Class II–Class III interface.33,34
 Only about 10% of HLA-Cw6 carriers develop psoriasis, and it has been estimated that PSORS1 accounts for only one-third of the variation in genetic liability to psoriasis.35 Therefore, it is highly likely that additional non-MHC genes are also involved. In addition to PSORS1, linkage studies have reported 18 potential susceptibility loci.17,36 However, many of these have proven difficult to replicate. After PSORS1, the second-most consistently replicated psoriasis susceptibility locus is PSORS2 (17q24-q25),37 with four independent studies providing confirmatory evidence (p < 0.01) in support of the original report.38–41 Other regions of putative linkage showing some evidence of reproducibility include PSORS4 (1q21.3),42,43 PSORS5 (3q21),44–46 PSORS8 (16q12-q13),47 and PSORS9 (4q28-q31).48,49
Only about 10% of HLA-Cw6 carriers develop psoriasis, and it has been estimated that PSORS1 accounts for only one-third of the variation in genetic liability to psoriasis.35 Therefore, it is highly likely that additional non-MHC genes are also involved. In addition to PSORS1, linkage studies have reported 18 potential susceptibility loci.17,36 However, many of these have proven difficult to replicate. After PSORS1, the second-most consistently replicated psoriasis susceptibility locus is PSORS2 (17q24-q25),37 with four independent studies providing confirmatory evidence (p < 0.01) in support of the original report.38–41 Other regions of putative linkage showing some evidence of reproducibility include PSORS4 (1q21.3),42,43 PSORS5 (3q21),44–46 PSORS8 (16q12-q13),47 and PSORS9 (4q28-q31).48,49
 It was reported that the PSORS2 locus contributes to psoriasis by influencing the expression of the SLC9A3R1, NAT1, and/or RAPTOR genes involved in immunologic regulation,50 but others have been unable to confirm these findings.51,52 Reports of two large families linked to this locus34,37 raise the possibility that more than one susceptibility gene may reside at PSORS2—one carrying a common disease allele with low penetrance, and the other carrying one or more rare alleles with high penetrance. The PSORS4 locus contains the epidermal differentiation complex (EDC). At least 58 genes involved in epidermal differentiation reside in the EDC, including the loricrin, involucrin, filaggrin, small proline-rich region (SPRR), S100, and late cornified envelope (LCE) genes.53 The PSORS5 locus (3q21) was originally linked to psoriasis in Swedish families;45 the evidence for replication is based on two association studies.45,46 The PSORS8 locus overlaps with a known susceptibility gene for Crohn disease (NOD2/CARD15) and has been implicated in psoriatic arthritis.47 The PSORS9 locus was originally identified in a Chinese population, but also provided some evidence for linkage in four other genome-wide linkage scans involving largely Caucasian populations.49
It was reported that the PSORS2 locus contributes to psoriasis by influencing the expression of the SLC9A3R1, NAT1, and/or RAPTOR genes involved in immunologic regulation,50 but others have been unable to confirm these findings.51,52 Reports of two large families linked to this locus34,37 raise the possibility that more than one susceptibility gene may reside at PSORS2—one carrying a common disease allele with low penetrance, and the other carrying one or more rare alleles with high penetrance. The PSORS4 locus contains the epidermal differentiation complex (EDC). At least 58 genes involved in epidermal differentiation reside in the EDC, including the loricrin, involucrin, filaggrin, small proline-rich region (SPRR), S100, and late cornified envelope (LCE) genes.53 The PSORS5 locus (3q21) was originally linked to psoriasis in Swedish families;45 the evidence for replication is based on two association studies.45,46 The PSORS8 locus overlaps with a known susceptibility gene for Crohn disease (NOD2/CARD15) and has been implicated in psoriatic arthritis.47 The PSORS9 locus was originally identified in a Chinese population, but also provided some evidence for linkage in four other genome-wide linkage scans involving largely Caucasian populations.49
 More recently, researchers studying psoriasis and other complex genetic disorders have turned to genome-wide association studies (GWAS) to identify susceptibility genes. In GWAS, disease allele frequencies are compared in cases and controls, rather than measuring the transmission of alleles through pedigrees as is done in linkage studies. Association is much more powerful than linkage in the search for common alleles contributing to polygenic diseases.16 The major disadvantage of genetic association studies is that their “effective range” is short, due to the many generations and numerous meiotic recombination events between the initial appearance of an ancient, disease-predisposing genetic variant and the present day. Therefore, hundreds of thousands of genetic markers are required to cover the genome in an association study.16 Fortunately, recently developed technologies allow 500,000 to 5,000,000 genetic markers, called single nucleotide polymorphisms (SNPs), to be typed at the same time. The identification of these SNPs allowed the development of the HapMap,54 which provides a dense map of SNP haplotypes (i.e., the SNP alleles found together on a single chromosome) from diverse ethnic populations. These resources allow geneticists to infer the genotypes of SNPs located between experimentally typed markers, a process called genotype imputation.55 Although the GWAS strategy entails a very large number of tests and therefore increases the risk of false-positive results, the ability to confirm the result in independent replication cohorts addresses this issue. Compounding the problem, it has become evident that the risk conferred by individual disease alleles is relatively small, meaning that thousands of cases and thousands of controls are typically required to carry out a well-powered GWAS. Fortunately, the prevalence of psoriasis (approximately 2% in Caucasians) is high enough that assembly of such large collections is feasible. With the advent of genome-wide association studies (GWAS), our knowledge of non-MHC genetic determinants of psoriasis susceptibility has advanced rapidly.56 At least 18 published non-MHC genetic loci meet strict criteria for genome-wide significance34,57–59 (Table 18-1). Of these, four [(1) IL12B, (2) IL23A, (3) IL23R, (4) TRAF3IP2] map within or near genes involved in the IL-23/Th17 axis.34,57,60,61 Additional regions of association contain genes whose products are involved in the control of Th1/Th2 polarization (IL4/IL13),34,63 in regulating NF-κB signaling (TNFAIP3, TNIP1, NFKBIA, and FBXL19),34,58,59,61,64 in MHC Class I antigen processing (ERAP1, PSMA6),59,65 in inflammatory dendritic cell function (NOS2), in interferon signaling (TYK2, IFIH1, IL28RA),61, 63–64 and in epidermal barrier formation (LCE3B/LCE3C).57,65 A report of increased defensin gene cluster copy number in psoriasis66 is of interest but remains to be confirmed. Later in this chapter, these findings are further examined in the context of the skin immune system (see Section “Psoriasis: Integrating Genetics and Immunology”).
More recently, researchers studying psoriasis and other complex genetic disorders have turned to genome-wide association studies (GWAS) to identify susceptibility genes. In GWAS, disease allele frequencies are compared in cases and controls, rather than measuring the transmission of alleles through pedigrees as is done in linkage studies. Association is much more powerful than linkage in the search for common alleles contributing to polygenic diseases.16 The major disadvantage of genetic association studies is that their “effective range” is short, due to the many generations and numerous meiotic recombination events between the initial appearance of an ancient, disease-predisposing genetic variant and the present day. Therefore, hundreds of thousands of genetic markers are required to cover the genome in an association study.16 Fortunately, recently developed technologies allow 500,000 to 5,000,000 genetic markers, called single nucleotide polymorphisms (SNPs), to be typed at the same time. The identification of these SNPs allowed the development of the HapMap,54 which provides a dense map of SNP haplotypes (i.e., the SNP alleles found together on a single chromosome) from diverse ethnic populations. These resources allow geneticists to infer the genotypes of SNPs located between experimentally typed markers, a process called genotype imputation.55 Although the GWAS strategy entails a very large number of tests and therefore increases the risk of false-positive results, the ability to confirm the result in independent replication cohorts addresses this issue. Compounding the problem, it has become evident that the risk conferred by individual disease alleles is relatively small, meaning that thousands of cases and thousands of controls are typically required to carry out a well-powered GWAS. Fortunately, the prevalence of psoriasis (approximately 2% in Caucasians) is high enough that assembly of such large collections is feasible. With the advent of genome-wide association studies (GWAS), our knowledge of non-MHC genetic determinants of psoriasis susceptibility has advanced rapidly.56 At least 18 published non-MHC genetic loci meet strict criteria for genome-wide significance34,57–59 (Table 18-1). Of these, four [(1) IL12B, (2) IL23A, (3) IL23R, (4) TRAF3IP2] map within or near genes involved in the IL-23/Th17 axis.34,57,60,61 Additional regions of association contain genes whose products are involved in the control of Th1/Th2 polarization (IL4/IL13),34,63 in regulating NF-κB signaling (TNFAIP3, TNIP1, NFKBIA, and FBXL19),34,58,59,61,64 in MHC Class I antigen processing (ERAP1, PSMA6),59,65 in inflammatory dendritic cell function (NOS2), in interferon signaling (TYK2, IFIH1, IL28RA),61, 63–64 and in epidermal barrier formation (LCE3B/LCE3C).57,65 A report of increased defensin gene cluster copy number in psoriasis66 is of interest but remains to be confirmed. Later in this chapter, these findings are further examined in the context of the skin immune system (see Section “Psoriasis: Integrating Genetics and Immunology”).
SNP ID (rs number)a | Chromosomal Band | Risk Allele (frequency in controls)b | Odds Ratiob | p-Valuec | Notable Gene(s) in Associated Region | Independently Confirmed | Reference |
|---|---|---|---|---|---|---|---|
rs12191877 | 6p21.33 | T (0.147) | 2.64 | <1 E-100 | HLA-C, CDSN | Y | Nair et al34 |
rs17728338 | 5q33.1 | A (0.054) | 1.59 | 1 E-20 | TNIP1 | Y | Nair et al34 |
rs2082412 | 5q33.3 | G (0.798) | 1.44 | 2 E-28 | IL12B | Y | |
rs33980500 | 6q21 | T (0.071) | 1.38 | 1 E-16 | TRAF3IP2 | Y | Ellinghaus et al58 |
rs4649203 | 1p36 | A (0.770) | 1.36 | 7 E-8 | IL28RA | N | Strange et al61 |
rs2066808 | 12q13.3 | A (0.932) | 1.34 | 1 E-09 | IL23A, STAT2 | Y | Nair et al34 |
rs17716942 | 2p24 | A (0.900) | 1.29 | 1 E-13 | IFIH1 | N | Strange et al61 |
rs20541 | 5q31.1 | G (0.790) | 1.27 | 5 E-15 | IL13, IL4 | N | Nair et al34 |
rs4085613 | 1q21.3 | C (0.421) | 1.27 | 7 E-30 | LCE3C, LCE3D | Y | Zhang et al57 |
rs495337 | 20q13.13 | C (0.551) | 1.25 | 1 E-08 | RNF114 | Y | Capon et al62 |
rs2431697 | 5q33.3 | C (0.177) | 1.19 | 1 E-08 | PTTG1 | N | Stuart et al59 |
rs4795067 | 17q11.2 | G (0.349) | 1.19 | 4 E-11 | NOS2 | Y | Stuart et al59 |
rs610604 | 6q23.3 | G (0.320) | 1.19 | 9 E-12 | TNFAIP3 | Y | Nair et al34 |
rs10782001 | 16p11.2 | G (0.368) | 1.16 | 9 E-10 | FBXL19 | N | Stuart et al64 |
rs12586317 | 14q13.2 | T (0.751) | 1.16 | 2 E-08 | NFKBIA, PSMA6 | Y | Stuart et al64 |
rs3751385 | 13q12.11 | T (0.478) | 1.14 | 2 E-10 | GJB2 | N | Stuart et al59 |
rs10088247 | 8p23.2 | C (0.183) | 1.13 | 5 E-09 | CSMD1 | N | Stuart et al59 |
rs2201841 | 1p31.3 | G (0.295) | 1.13 | 3 E-08 | IL23R | Y | Nair et al34 |
rs151823 | 5q15 | A (0.495) | 1.12 | 9 E-09 | ERAP1 | Y | Stuart et al59 |
rs514315 | 18q22.1 | T (0.742) | 1.12 | 6 E-09 | SERPINB8 | N | Stuart et al59 |
rs9304742 | 19q13.41 | T (0.649) | 1.11 | 2 E-09 | ZNF816A | N | Stuart et al59 |
NAd | 8p23.1 | NAd | NAd | 3 E-08 | β-defensin genes | N | Hollox et al66 |
 An interesting feature of the GWAS results obtained thus far in psoriasis and other complex genetic disorders is that the SNP allele conferring risk of disease is often more common than the nonrisk in allele not only in cases but also in normal controls. In some cases, the disease allele may be ancestral, as is the case for lactose intolerance. Alternatively, the disease allele may be beneficial in certain contexts (i.e., defense against pathogens), as is the case for hemoglobinopathies increasing resistance to malaria, or at least selectively neutral with respect to reproduction. It is also possible that the rare variant may actually encode a protective function. Finally, the actual functional variant may be rare, but carried on a common haplotype tagged by the observed variant. Fine mapping and functional studies of psoriasis and other complex genetic disorders are in their early stages. The outcome of these studies should help to distinguish between these possibilities.
An interesting feature of the GWAS results obtained thus far in psoriasis and other complex genetic disorders is that the SNP allele conferring risk of disease is often more common than the nonrisk in allele not only in cases but also in normal controls. In some cases, the disease allele may be ancestral, as is the case for lactose intolerance. Alternatively, the disease allele may be beneficial in certain contexts (i.e., defense against pathogens), as is the case for hemoglobinopathies increasing resistance to malaria, or at least selectively neutral with respect to reproduction. It is also possible that the rare variant may actually encode a protective function. Finally, the actual functional variant may be rare, but carried on a common haplotype tagged by the observed variant. Fine mapping and functional studies of psoriasis and other complex genetic disorders are in their early stages. The outcome of these studies should help to distinguish between these possibilities.
Detailed light, electron microscopic, immunohistochemical, and molecular studies of involved and uninvolved skin of both newly appearing and established psoriatic lesions provide a useful framework for inferring cause-and-effect relationships between the many cellular events that take place in a psoriatic lesion. They are illustrated schematically in Fig. 18-1 and with actual photomicrographs in Fig. 18-2.
Figure 18-1

Development of psoriatic lesions. This figure depicts the transition from normal skin to fully developed lesion described in the text. Normal skin from a healthy individual (panel A) contains epidermal Langerhans cells, scattered immature dendritic cells (D), and skin-homing memory T cells (T) in the dermis. Normal-appearing skin from a psoriatic individual (panel B) manifests slight capillary dilatation and curvature, and a slight increase in the numbers of dermal mononuclear cells and mast cells (M). A slight increase in epidermal thickness is usually present. In chronic plaque psoriasis, the intensity of these changes may depend on distance from an established lesion. The transition zone of a developing lesion (panel C) is characterized by progressive increases in capillary dilatation and tortuosity, numbers of mast cells, macrophages (MP), and T cells, and mast cell degranulation (small arrows). In the epidermis, there is increasing thickness with increasingly prominent rete pegs, widening of the extracellular spaces, transient dyskeratosis, spotty loss of the granular layer, and parakeratosis. Langerhans cells (L) begin to exit the epidermis, and inflammatory dendritic epidermal cells (I) and CD8+ T cells (8) begin to enter the epidermis. The fully developed lesion (panel D) is characterized by fully developed capillary dilatation and tortuosity with a tenfold increase in blood flow, numerous macrophages underlying the basement membrane, and increased numbers of dermal T cells (mainly CD4+) making contact with maturing dermal dendritic cells (D). The epidermis of the mature lesion manifests markedly increased (approximately tenfold) keratinocyte hyperproliferation extending to the lower suprabasal layers, marked but not necessarily uniform loss of the granular layer with overlying compaction of the stratum corneum and parakeratosis, increased numbers of CD8+ T cells, and accumulation of neutrophils in the stratum corneum (Munro’s microabscesses).
Figure 18-2
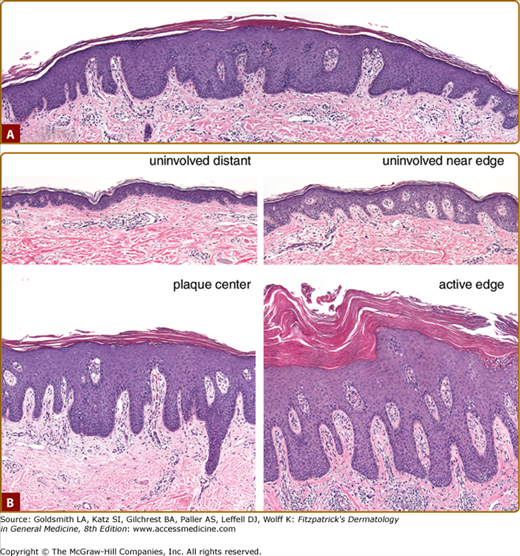
Histopathology of psoriasis. A. Pinpoint papule of psoriasis. In the transition from the edges to the center of the lesion, note progressive thickening of epidermis with elongation of rete pegs, increasing dilatation and tortuosity of vessels, and increasing mononuclear cell infiltrate. Also note the transition from basket-weave to compact stratum corneum with loss of granular layer in the center of the lesion. (4-mm punch biopsy, hematoxylin and eosin, scale bar = 100 μM.) B. Comparison of uninvolved versus involved skin. Four 4-mm biopsies were taken from the same individual sampled in A on the same day. “Uninvolved distant” skin was taken from the upper back 30 cm from the nearest visible lesion of psoriasis. “Uninvolved near edge” skin was taken 0.5 cm from the edge of a 20-cm plaque, which had been present for several years, according to the patient. “Center plaque” skin was taken from a relatively inactive (less red and scaly) area in the center of this plaque. “Involved edge” skin was taken from an active (more red and scaly) area about 1 cm inside the edge of the same plaque. In comparing “uninvolved distant” to “uninvolved near edge” skin, note that the latter manifests increased thickness and early elongation of the rete pegs, dilatation and early tortuosity of blood vessels, and increased numbers of mononuclear cells in the upper dermis, many of which are in a perivascular location. In this patient, “uninvolved near edge” skin also manifests an increased frequency of dyskeratotic keratinocytes, a finding that has been noted previously at the periphery of psoriatic lesions.53 In comparing less active to more active areas of the plaque, note that the more active area manifests increased dermal mononuclear infiltrate, increased hyperkeratosis and parakeratosis, and Munro’s microabscesses. (4-mm punch biopsies, hematoxylin, and eosin, scale bar = 100 μM.)
The normal-appearing skin of psoriatic patients has long been known to manifest subclinical morphologic and biochemical changes, particularly involving lipid biosynthesis.67,68 These changes were predominantly found in the stratum corneum and included changes in the levels and composition of phospholipids, free α-amino acids, hydrolytic enzymes, and several dehydrogenases. These changes led to the use of the term “histochemical parakeratosis” to describe these findings.67 Much more recent studies using microarray technology to search for differences in gene expression between normal and uninvolved psoriatic skin have identified groups of coordinately regulated genes involved in lipid biosynthesis and innate immune defense.69
In the initial pinhead-sized macular lesions there is marked edema, and mononuclear cell infiltrates are found in the upper dermis.70 These findings are usually confined to the area of one or two papillae. The overlying epidermis soon becomes spongiotic, with focal loss of the granular layer. The venules in the upper dermis dilate and become surrounded by a mononuclear cell infiltrate.67 Similar findings have been described in early macules and papules of psoriasis71 and in clinically normal-appearing skin 2–4 cm away from any active lesion in patients undergoing an acute flare of guttate psoriasis.72
Studies of the clinical margins of somewhat larger lesions (0.5–1.0 cm) reveal an approximately 50% increase in epidermal thickening in the “normal-appearing” skin immediately adjacent to lesions.67 There is a large increase in the metabolic activity of epidermal cells, including the stratum corneum, increased DNA synthesis, an increased number of mast cells and dermal macrophages, and increased mast cell degranulation.67,73,74 Subsequent studies revealed increased numbers of dermal T cells75 and dendritic cells (DCs)76 in both uninvolved and involved psoriatic skin relative to normal skin. Toward the center of the lesion, a “marginal zone” can be identified, with increasing band-like epidermal thickness, increasing parakeratosis and capillary elongation, and perivascular infiltration of lymphocytes and macrophages, without exudation into the epidermis. More centrally, rete ridges begin to develop in the marginal zone, before finally transitioning into the fully developed psoriatic plaque. Squamous cells manifest enlarged extracellular spaces with only a few desmosomal connections. Parakeratosis is typically mounded or spotty.
Mature lesions of psoriasis are characterized by uniform elongation of rete ridges, with thinning of the epidermis overlying the dermal papillae.67,71 Epidermal mass is increased three to five times, and many more mitoses are observed, frequently above the basal layer. About 10% of basal keratinocytes are cycling in normal skin, whereas this value rises to 100% in lesional psoriatic skin.77 Widening of the extracellular spaces between keratinocytes persists but is less prominent than in developing lesions and is more uniform than the typical spongiosis of eczematous skin lesions. The tips of the rete ridges are often clubbed or fused with adjacent ones, with thin, elongated, edematous papillae containing dilated, tortuous capillaries. Parakeratosis, with accompanying loss of the granular layer, is often horizontally confluent but may alternate with orthokeratosis,78 and hyperkeratosis is more extensive than in the transitional zone. The inflammatory infiltrate around the blood vessels in the papillary dermis becomes more intense but still consists of lymphocytes, macrophages, DCs, and mast cells. Unlike the initial lesion and the transitional zone, lymphocytes are observed in the epidermis of the mature lesion. Neutrophils exit from the tips of a subset of dermal capillaries (the “squirting papillae”), leading to their accumulation in the overlying parakeratotic stratum corneum (Munro’s microabscesses) and, less frequently, in the spinous layer (spongiform pustules of Kogoj). Collections of serum can also be seen in the epidermis and the stratum corneum.67,71
In 1984, it was demonstrated that the eruption of psoriatic skin lesions coincided with epidermal influx and activation of T cells,75 and shortly thereafter it was further shown that resolution of psoriasis during phototherapy was preceded by depletion of T cells, predominantly from the epidermis.81 Several studies found CsA to be highly effective in psoriasis,82,83 and this effect was demonstrated to be primarily through blockade of T cells rather than keratinocytes.84 Furthermore, psoriasis has been triggered or cured by bone marrow transplantation, depending on whether the donor or the host was psoriatic.85,86 The role of T cells in psoriasis was functionally demonstrated in 1996 when it was shown that the psoriasis process could be induced by injecting activated autologous T cells into uninvolved psoriatic skin transplanted onto severe combined immunodeficient mice.87 Available data indicate that the T-cell responses are antigen-specific rather than mediated by superantigens, as clonal populations of both CD4+ and CD8+ T cells have consistently been identified in psoriatic lesions.88–91 However, most of the T cells in a psoriatic lesion are not clonally expanded and may accumulate in response to the cytokine environment of the lesion. There is virtually no evidence for B-cell involvement or antibody-mediated processes in psoriasis.
The best-characterized T cells are the CD4+ and CD8+ subsets. Predominantly of the memory phenotype (CD45RO+), these cells express the cutaneous lymphocyte antigen (CLA), a ligand for E-selectin, which is selectively expressed on skin capillaries and therefore provides them with access to the skin.92 CD8+ T cells are predominantly located in the epidermis, whereas CD4+ T cells are predominantly located in the upper dermis.93,94 The cytokine profile of psoriatic lesions is rich in interferon (IFN)-γ,95 indicative of T helper 1 (Th1) polarization of CD4+ cells, and T cytotoxic 1 (Tc1) polarization of CD8+ cells96 (Fig. 18-3). Two other subsets of CD4+ T cells, stimulated by IL-23 and characterized by production of IL-17 (Th17 cells) and/or IL-22 (Th22 cells), are also found in psoriatic lesions and have been shown to play a major role in maintaining chronic inflammation in psoriasis97,98 as well as other autoinflammatory conditions.99–101 While the majority of CD4 T cells are Th1, about 20% of them produce IL-17 (Th17) and ∼15% produce IL-22 (Th22).98 Similarly, CD8+ epidermal T cells producing IFN-γ (Tc1), IL-17 (Tc17), and IL-22 (Tc22) are found in psoriasis.98 These T-cell subsets have considerable functional plasticity and conversions of Tc17 to Tc1102 and Th17 to Th1103–105 have been described. In mice most Th17 cells also elaborate IL-22, which mediates dermal inflammation and epidermal hyperplasia after intracutaneous injection of IL-23.106 However, in humans, this overlap is much less pronounced, with largely distinct populations of Th17 and Th22 cells.98,107–109
Figure 18-3

The cytokine network in psoriasis. IFN-γ is produced by Th1 cells, and TNF-α is produced by activated T-cells and DCs. IFN-γ amplifies the production of IL-23 by DC. In turn, IL-23 maintains and expands subsets of CD4+ T cells, called Th17 and Th22 cells, which are characterized by production of IL-17 and IL-22, respectively. CD8+ T cells are predominantly found in the epidermis, and their entry into the epidermis is necessary for lesion development. IL-17, TNF-α, IFN-γ, and IL-22 synergistically promote activation of the innate keratinocyte defense response involving secretion of antimicrobial peptides such as human-β-defensin 2 (hBD-2), IL-8 and other chemokines, and growth factors such as TGF-α, AREG, IL-19, and IL-20. Keratinocytes also produce IL-7 and IL-15, which influence the survival and turnover of CD8+ T cells, and IL-18, which via IL-12 causes DC to further increase the production of IFN-γ by T-cells.
Regulatory T cells suppress immune responses in an antigen-specific fashion, and are responsible not only for downregulating successful responses to pathogens but also for the maintenance of immunologic tolerance.110 Several different populations of regulatory T cells (T-regs) exist but the best characterized one is the CD4+ CD25+ subset.111 A recent study of this subset in psoriasis demonstrated impaired inhibitory function and failure to suppress effector T-cell proliferation,112 possibly due to a tissue environment rich in IL-6 produced by endothelial, dendritic, and Th17 cells.113
Natural killer T cells (NKT cells) are a heterogeneous subpopulation of T lymphocytes defined by coexpression of the T-cell receptor (TCR) and natural killer (NK) lineage markers such as CD16, CD56, CD57, CD94, and CD161. Unlike conventional T cells, NKT cells recognize glycolipid antigens in the context of the MHC Class I-like antigen-presenting molecule CD1d. NKT cells constitute only a small fraction of lymphocytes. Nevertheless, their ability to rapidly secrete large amounts of cytokines, including IFN-γ, IL-4, IL-2, IL-5, IL-10, IL-13, IL-17, and TNF-α, positions them as potentially important regulators of T-cell differentiation at sites of inflammation. While NKT cells are increased in psoriatic lesions relative to uninvolved or normal skin, their precise role in psoriasis remains unclear.114
Like NKT cells, NK cells are major producers of IFN-γ and serve as a bridge between innate and acquired immunity. Unlike NKT cells, NK cells do not express the T-cell receptor. NK cells are present in psoriasis,115,116 and can trigger the formation of psoriasis lesions in a xenograft model system.117 NK cells are regulated in part by killer immunoglobulin-like receptors (KIRs), which recognize HLA-C and other MHC Class I molecules. KIRs are a family of ∼15 closely linked genes located on chromosome 19q13.4,118 some of which stimulate and others of which inhibit NK cell activation. KIR genes have been associated with psoriasis119–121 and psoriatic arthritis.122,123 It has been proposed that susceptibility to psoriatic arthritis is determined by the overall balance of activating and inhibitory genotypes.121,124 Although it is attractive to speculate that the association of psoriasis with HLA-Cw6 might reflect a KIR-mediated dysregulation of NK cells, it is known that a number of other HLA-C protein alleles recognize the same inhibitory receptor (KIR2DL1), including HLA-Cw2, HLA-Cw4, HLA-Cw5, HLA-Cw15, and HLA-Cw17. Thus, it is not straightforward to explain the action of HLA-Cw6 in psoriasis on the basis of KIR recognition alone.
Treatments directed primarily against key costimulatory molecules expressed by “professional” antigen-presenting DCs markedly improve psoriasis.79 This suggests that T cells in psoriatic lesions are in constant communication with DCs, which have a role in both the priming of adaptive immune responses and the induction of self-tolerance125 (see Chapter 10). Several subsets of DCs have been defined, and many of these are found in markedly increased numbers within psoriatic lesions.125–128 Although DCs are believed to be central to the pathogenesis of psoriasis, the specific role of each subset is still somewhat unclear.
Usually defined by a Langerin+, CD1a+ surface phenotype, Langerhans cells (LCs) are considered to be immature DCs (iDCs). LCs have a well-defined role as antigen-presenting cells (APCs) in contact dermatitis,129 but their role in psoriasis is currently somewhat unclear. While the density of LC is decreased in lesional psoriasis in terms of cells per unit area,126,130 the number of LC per unit length of epidermis is similar in normal, uninvolved, and lesional skin.128 DCs lacking the characteristic Birbeck granule but positive for the maturation molecule DC-LAMP found in the dermis of psoriatic lesions could be derived from epidermal or dermal iDC.131 Recently, LC have been shown to preferentially drive Th22 differentiation, relative to dermal DC.132 Interestingly, migration of LCs in response to inflammatory cytokines is markedly impaired in uninvolved psoriatic epidermis relative to normal skin,133 especially in type I (early onset) psoriasis.134
Dermal DCs do not express activation markers in resting normal skin and in that context can be considered as another type of iDC that is similar to myeloid iDCs found in other tissues.128,135 Immunophenotyping studies have revealed that the population of dermal DCs is quite complex, and that psoriasis lesions demonstrate a marked increase in the number and maturation state of dermal DC.126,136,137 Identified initially by strong expression of MHC Class II and/or factor XIIIa,138 it is now appreciated that factor XIIIa+ cells are macrophages rather than DCs, and that the most reliable marker for myeloid-derived dermal DC is CD11c.139 There appears to be three types of myeloid-derived (CD11c+) DCs in psoriasis lesions.128,139,140 The first is the population of “resident” dermal DCs that are also seen in normal skin. These CD11c+/CD1c+ DC account for about 10%–15% of DCs in psoriasis lesions. These cells are relatively more mature than inflammatory DCs (see Section “Inflammatory Dendritic Epidermal Cells”), but less so than fully mature DCs. The second population comprises mature DCs that are marked by DC-LAMP or CD83. The DC-LAMP+ DCs form large aggregates with T cells in the dermis, whereas the CD83+ DCs are more diffuse. It has been suggested that fully mature DCs may be the sustaining force for chronic T-cell activation in skin and the characteristics of these cells are very similar to those in lymph nodes.128,139–141 The third population of myeloid DCs are the inflammatory DCs, which are CD11c+/CD1c−, and are less mature than the resident CD1c+ subset. These cells make IL-23 and probably help drive Th17 differentiation.142 About 80%–90% of DCs in psoriasis lesions are these relatively immature, inflammatory DCs and, interestingly, the total number of these cells can exceed the number of T cells in lesions. A subset of these inflammatory DC express high levels of TNF-α and iNOS, and by analogy to similar if not identical cells in mice, have been called “TIP-DC” (for TNF-α and iNOS-producing DC). Consistent with our recent genetic findings,64 TIP-DCs are increased up to 30-fold in psoriatic lesions.128 There appears to be substantial plasticity in this population of cells, as the cytokine milieu in atopic dermatitis promotes the emergence of dermal DC that chemotactically attract a different subset of T cells than those found in psoriasis.143
Thought to be either monocyte-derived iDCs,144 or a variant of inflammatory myeloid DC that migrate into the epidermis,145 inflammatory dendritic epidermal cells (IDECs) are distinguished from LCs by the lack of Birbeck granules and lower expression of CD1a. Unlike LCs, IDECs are nearly absent in normal skin, and their numbers are markedly increased in the epidermis of active psoriasis lesions, as well as a large number of other inflammatory dermatoses.126,130
Plasmacytoid DCs (pDCs) are inefficient presenters of antigens to T cells. However, they regulate inflammation and link innate with adaptive immunity, producing large amounts of IFN-α when activated146 (see Chapter 10). Absent from normal skin, pDCs are significantly increased in both uninvolved and involved psoriatic skin, but activated only in involved skin.126,147 Interestingly, inhibition of pDCs was shown to prevent development of psoriasis in a mouse xenograft model.147 Conversely, imiquimod, which has been reported to exacerbate psoriasis,148 likely acts through this type I IFN system by binding to Toll-like receptor 7 on pDCs.149 Although IFN-α appears to have a role in psoriatic lesional development and exacerbations,147 its role in stable chronic plaque psoriasis has been questioned.150
Mast cells have long been observed in initial and developing psoriasis lesions,67 with prominent mast cell degranulation in both eruptive psoriasis72 and in lesions reappearing after discontinuation of topical corticosteroid suppression.74 Interestingly, skin-derived mast cell release of preformed and newly synthesized mediators is potently suppressed byCsA and tacrolimus,151 suggesting that the antipsoriatic effects of these compounds could be mediated by mast cells as well as T cells. Recently, mast cells have been shown to be a major source of IL-17 production in both rheumatoid arthritis synovium152 and in psoriatic lesions.153
Macrophages are prominent in initial and developing psoriasis lesions.67 CD163 has recently been shown to be a reliable marker for skin-derived macrophages,139 and as mentioned earlier, these cells also express Factor XIIIa.154 A population of CD11c−, CD1a+, CD68+ macrophages is found scattered just under the basement membrane, subadjacent to proliferating keratinocytes expressing the macrophage chemokine MCP-1 (CCL2).155–157 These phagocytically active cells could be involved in generating fenestrations (holes) in the epidermal basement membrane.158 Recent studies in two different mouse models of psoriasis, one dependent on and the other independent of T cells, showed that selective elimination of macrophages led to prompt improvement of lesions. These findings suggest that macrophages may play a key role in the pathogenesis of psoriasis, at least in part via production of tumor necrosis factor (TNF)-α, iNOS, and IL-23.154,159–161
Although neutrophils are commonly seen in the upper epidermis of psoriatic lesions, they appear late during the development of lesions, their number is quite variable, and their role in the pathogenesis of psoriasis is unclear. Studies in one of the same mouse models used to implicate macrophages indicate that neutrophils are probably unnecessary for lesional development.161
As detailed below, keratinocytes are a major producer of proinflammatory cytokines, chemokines, and growth factors,162 as well as other inflammatory mediators such as eicosanoids163 and mediators of innate immunity such as cathelicidins, defensins, and S100 proteins.164 Psoriatic keratinocytes are engaged in an alternative pathway of keratinocyte differentiation called regenerative maturation.165 Regenerative maturation is activated in response to immunologic stimulation in psoriasis,166 but the mechanism by which this occurs is presently unknown.
Other cell types, such as endothelial cells and fibroblasts, are also likely to be participants in the pathogenic process. Endothelial cells are strongly activated in developing and mature lesions of psoriasis67,71 and in addition to delivering a tenfold increase in blood flow to the lesion, they play a major role in controlling the flux of leukocytes and serum proteins into psoriatic tissue.167–169 Fibroblasts support keratinocyte proliferation in a paracrine manner,170 and this process is exaggerated in psoriasis.171 Fibroblasts produce many chemotactic factors and support the migration of T cells out of psoriatic lesions.172 Thus, fibroblasts may also be intimately involved in psoriasis by directing the localization of T cells.
The cytokine network in psoriasis is extremely complex, involving the actions and interactions of multiple cytokines, chemokines, and growth factors, and their receptors in addition to other mediators produced by multiple cell types. Combinations of cytokines and growth factors can result in effects that are not seen when these factors are studied individually. For example, T-cell clones isolated from psoriatic skin lesions are able to promote keratinocyte proliferation in an IFN-γ-dependent manner,173,174 but by itself, IFN-γ has an antiproliferative effect on cultured keratinocytes.175
The most prominent cytokines currently thought to be involved in the pathogenesis of psoriasis are summarized in Fig. 18-3. Besides IFN-γ,96,176 a plethora of cytokines and chemokines are upregulated in psoriasis, including the cytokines TNF-α,177 IL-2,95 IL-6,178 IL-8,179 IL-15,180 IL-17,181 IL-18,182 IL-19, IL-20, IL-22,183 IL-21,184 IL-23,97,100,101 and the chemokines MIG/CXCL9, IP-10/CXCL10, I-TAC/CXCL11, and MIP-3α/CCL20.7,185 More complex abnormalities have been observed for other immunomodulatory cytokines and their receptors, including IL-1 and TGF-β.186–188 IL-23 is currently believed to play a central role in the pathogenesis of psoriasis through its role in maintaining and expanding specific subsets of CD4+ T cells characterized by production of IL-17 (Th17)189 and IL-22 (Th22).106 IFN-γ plays a role in amplifying this process by stimulating antigen-presenting cells to produce more IL-23.190 The entry of activated, cytokine secreting, CD8+ T cells into the epidermis promotes epidermal hyperplasia191 and activation of keratinocyte innate defense response with resulting production of antimicrobial peptides, cytokines, chemokines, and growth factors (see Fig. 18-3). The other major cytokine producers in psoriatic lesions are DC and macrophages,154 with additional contributions from mast cells, neutrophils, and endothelial cells. Overall, this creates a highly complex network of inflammatory signals between the main cellular participants.
In addition to cytokines and chemokines, several mediators of innate immunity are abnormally expressed in psoriasis.164 Prominent among the innate immune mediators are the antimicrobial peptides human β-defensin-2 (hBD-2) and cathelicidin (LL-37), both of which are much more highly overexpressed in psoriasis than in atopic dermatitis.192,193 Notably, expression of HBD-2 and LL-37 is increased in response to proinflammatory and type I cytokines (TNF-γ, IL-1, and IFN-γ) and suppressed by type II cytokines (IL-4, IL-10, and IL-13).194 These differences in antimicrobial peptide expression help to explain why approximately 30% of patients with atopic dermatitis have bacterial or viral infections, as opposed to only 7% of psoriasis patients, even though both conditions have a defective skin barrier.195 They may also explain why psoriasis patients, though frequently colonized with Staphylococcus aureus, are not markedly improved by antibiotic treatment, whereas atopic dermatitis patients often obtain dramatic benefit from antibiotic therapy. The S100 proteins are a large family of dimeric low-molecular-weight proteins that bind calcium and other divalent cations. S100A2, S100A7 (psoriasin), and the S100A8/A9 heterodimer (calprotectin) are markedly overexpressed in psoriasis lesions.196 These proteins exert chemotactic and antimicrobial activity, the latter through sequestration of zinc ions,197,198 and have been shown to function as TLR4 ligands on CD8+ T cells, upregulating IL-17 expression and inducing autoimmunity.199 Nitric oxide is produced in large amounts by DCs in psoriasis, triggering multiple signal transduction events.137 Finally, the complement component C5a is a potent chemoattractant for neutrophils and may contribute to the accumulation of neutrophils in the stratum corneum of psoriasis.200 Interestingly, it is also the most potent chemoattractant for DCs in psoriatic scale extracts.201 Many of these mediators are regulated in response to Toll-like receptors, providing a mechanism whereby the innate immune system can rapidly recognize a wide variety of pathogens according to their pathogen-associated molecular patterns202 (see Chapter 10).
The role of eicosanoids in psoriasis remains unclear.203 Levels of free arachidonic acid, leukotriene B4, 12-hydroxyeicosatetraenoic acid, and 15-hydroxyeicosatetraenoic acid are markedly increased in lesional skin, whereas levels of prostaglandins E and F2α are increased less than twofold.
Multiple growth factors are overexpressed in psoriasis.204 Members of the epidermal growth factor (EGF) family induce their own production in keratinocytes, including transforming growth factor-α, amphiregulin (AREG), and heparin-binding EGF-like growth factor.205 Studies in xenografted mice found a reduction in epidermal hyperplasia after antibody-mediated neutralization of AREG.206 Activation of the EGF receptor stimulates keratinocyte production of vascular endothelial growth factor (VEGF),167 perhaps accounting for the long-standing observation that the angiogenic properties of normal and psoriatic skin associate with the epidermis.207 Nerve growth factor (NGF) is also overexpressed by keratinocytes in psoriatic skin, and NGF receptors are increased in the peripheral nerves of lesional skin. Psoriasis has been shown to clear in denervated skin,208 and psoriasis improved after NGF blockade in xenografted mice.209 Moreover, direct connections have been documented between terminal nerve fibers and DCs in the skin, and neuropeptides have been shown to modulate DC function.210 Paracrine growth factors produced outside the epidermal compartment may also play an important role in stimulating epidermal hyperplasia in psoriasis, including insulin-like growth factor-1211 and keratinocyte growth factor.212
The psoriatic lesion is characterized by marked overexpression of multiple classes of proteinases by both keratinocytes and leukocytes. Metalloproteinases release TNF-α, several EGF-like growth factors, and many other cytokines and growth factors from their membrane-anchored precursors.213 Leukocyte-derived elastase has also been implicated in the release of EGF-like growth factors.214 Serine proteases directly activate protease-activated receptors.215 Each of these mechanisms may contribute to stimulation of keratinocyte proliferation. The protease inhibitors elafin, serpinB3, and serpinB13 (hurpin) are among the most markedly overexpressed genes in psoriatic lesions,216,217 suggesting that homeostatic mechanisms are strongly engaged in an effort to regulate the proteolytic environment of psoriatic lesions.
Several observations suggest an early role for α5 integrins and their ligand fibronectin in psoriasis. Fibronectin is increased in psoriatic epidermis,218 and it has been suggested that fibronectin gains access to the epidermis via fenestrations in the epidermal basement membrane.219 Fibronectin receptors (α5 integrins) are only weakly expressed in normal skin, but are strongly expressed in uninvolved as well as involved psoriatic skin,219 and fibronectin selectively increases the proliferation of keratinocytes cultured from uninvolved psoriatic skin.219 Receptors for collagen and laminin-5 (α2β1 and α3β1 integrins, respectively) are confined to the basal layer of normal skin but are strongly expressed in suprabasal keratinocytes as part of the regenerative maturation program.220 Interestingly, forced expression of β1 integrins in the suprabasal compartment results in epidermal hyperplasia.221 Finally, the entry and retention of CD8+ T cells into the epidermis requires binding of type IV collagen by α1β1 integrin and binding of E-cadherin by αEβ7 integrin, respectively.191,222
As would be expected given this plethora of intercellular signaling alterations, multiple signal transduction mechanisms are dysregulated in psoriatic epidermis, including receptor tyrosine kinase, mitogen-activated protein kinase, Akt, STAT, Src family kinase, Wnt,223 and NF-κB pathways. These abnormalities affect immunocyte activation and trafficking as well as keratinocyte responses of proliferation, differentiation, and survival. As described below, many of the susceptibility variants associated with psoriasis have a role in regulating these signaling pathways, particularly the NF-κB pathway.34,64 This is a challenging area of research, as signal transduction experiments are typically conducted on cell lines in culture, whereas the phenotype of psoriasis requires intercellular interactions and differentiation conditions that can only be obtained in vivo. Animal models are helping to define signal transduction abnormalities at a functional level. Space does not allow detailed consideration of these pathways in psoriasis, nor of the animal models being used to study them. Interested readers are referred to several reviews for details.50,188,224–226
(Fig. 18-4). As has been made clear by detailed fine mapping, genetic linkage, and association studies, HLA-C is by far the major genetic risk factor for psoriasis.30,34,35,38,57 Because it presents antigens to CD8+ T cells, HLA-Cw6 is an excellent candidate for functional involvement in psoriasis. Psoriasis has long been known to be triggered by streptococcal pharyngitis, and is the only infection that has been shown to trigger psoriasis in a prospective cohort study.227 Because tonsillar T cells are cutaneous lymphoid antigen (CLA)-positive and recognize activated skin endothelium228 they can traffic into the skin, explaining why the same CLA-positive T-cell clones are found in the tonsils and in the lesional skin of psoriatic patients.229 CD8+ T cells comprise at least 80% of the T cells in the epidermis of psoriatic lesions,230 and epidermal invasion correlates with lesional development.94,231,232 CD8+ T cells selectively traffic to the epidermis because they express integrin α1β1 (also known as VLA-1), which binds to type IV basement membrane collagen,191 as well as integrin αEβ7, which binds to keratinocyte E-cadherin.222 Once in the epidermis, CD8+ T cells “interrogate” peptides bound to HLA-Cw6 on the surface of dendritic APCs and/or keratinocytes. In normal immune responses, CD4+ T cells provide critical help in processing and presentation of intracellular viral components and tumor antigens, in a process called cross-priming. While CD4+ and CD8+ memory T cells can traffic between the skin and lymph nodes and blood, increasing evidence suggest that they spend most of their time in the skin itself.233 This may help to account for the characteristic distribution of psoriatic plaques, which tend to recur in the same places after therapeutic or spontaneous improvement.
Figure 18-4

Proposed role of HLA-Cw6 in the pathogenesis of psoriasis. Antigen (Ag) in the binding pocket of HLA-Cw6 interacts with a T-cell receptor. The role of HLA-Cw6 in psoriasis is likely to be twofold. HLA-Cw6 is active in cross-presenting peptides on the surface of dendritic cells, allowing activation and clonal expansion of antigen-specific CD8+ T cells. This process is dependent on CD4+ T-cell help for cross-presentation of intracellular antigens and is likely to happen both in the dermis (activation of memory resident T cells) and local lymph nodes (activation of naive T cells). Subsequently, the CD8+ T cells are able to migrate into the epidermis where they encounter HLA-Cw6 on the surface of the keratinocytes presenting those same pathogenic peptides. Activated CD8+ T-cells may recognize peptides presented by HLA-Cw6 on keratinocyte cell surface. Because these T-cells express perforin, they could directly damage keratinocytes in the traditional cytotoxic manner.435 Activated CD8+ T cells could also trigger the local release soluble factors, including cytokines, chemokines, eicosanoids, and/or innate immune mediators, which could further increase local inflammation and stimulate keratinocyte proliferation.173 In response to either insult, keratinocytes could respond by elaborating autocrine growth factors such as TGF-α and AREG, thereby encouraging their own proliferation and survival.436
As mentioned earlier, there is a very strong association between HLA-Cw6 and guttate psoriasis. This form of psoriasis is often self-limiting5,234 but can progress to chronic plaque psoriasis, which has a fluctuating inflammatory course in the absence of ongoing streptococcal infection. The transition from guttate to chronic plaque psoriasis likely reflects a transition from a self-limited cutaneous immune reaction triggered by streptococci encountered in the tonsils during a guttate flare, to a persistent and inappropriate immune reaction directed against host proteins in chronic plaque disease.235 The mechanisms by which normal immunologic tolerance of self-proteins is broken during this transition remain to be elucidated.
 What Happens during a Guttate Flare?
What Happens during a Guttate Flare? During a guttate flare, we speculate that T cells respond to streptococcal infection by proliferating, differentiating into an effector/memory phenotype, and acquiring skin-homing capability. On entering the skin, these cells encounter a locally activated dermal environment characterized by capillary dilatation and edema.71,72,74,236 This environment may be fostered by focal mast cell degranulation and macrophage activation, with release of TNF-α leading to induction of adhesion molecules on the endothelial cell surface, facilitating entry of T cells into the dermis. The mechanisms that trigger the activation of these mast cells and macrophages are presently unknown. At this point in lesional evolution, epidermal changes are subtle but include an increase in keratinocyte DNA synthesis, widened extracellular spaces between keratinocytes, and biochemical alterations in the stratum corneum indicative of altered differentiation, despite a lack of visible parakeratosis.67 The mechanisms by which these epidermal changes are evoked remain unknown but could involve the generation of basement membrane fenestrations by macrophage-derived proteases,156,157 allowing permeation of fibronectin and even cytokine-laden mast cell granules73 into the epidermal compartment. The former event stimulates keratinocyte proliferation,219 whereas the latter could provoke the epidermal spongiosis characteristic of very early psoriatic lesions.73
During a guttate flare, we speculate that T cells respond to streptococcal infection by proliferating, differentiating into an effector/memory phenotype, and acquiring skin-homing capability. On entering the skin, these cells encounter a locally activated dermal environment characterized by capillary dilatation and edema.71,72,74,236 This environment may be fostered by focal mast cell degranulation and macrophage activation, with release of TNF-α leading to induction of adhesion molecules on the endothelial cell surface, facilitating entry of T cells into the dermis. The mechanisms that trigger the activation of these mast cells and macrophages are presently unknown. At this point in lesional evolution, epidermal changes are subtle but include an increase in keratinocyte DNA synthesis, widened extracellular spaces between keratinocytes, and biochemical alterations in the stratum corneum indicative of altered differentiation, despite a lack of visible parakeratosis.67 The mechanisms by which these epidermal changes are evoked remain unknown but could involve the generation of basement membrane fenestrations by macrophage-derived proteases,156,157 allowing permeation of fibronectin and even cytokine-laden mast cell granules73 into the epidermal compartment. The former event stimulates keratinocyte proliferation,219 whereas the latter could provoke the epidermal spongiosis characteristic of very early psoriatic lesions.73
 What Is (Are) the Psoriasis Autoantigen(s)?
What Is (Are) the Psoriasis Autoantigen(s)? If psoriasis is mediated by antigen-specific T cells, then what are the T cells reacting against? In guttate psoriasis, streptococcal M protein is a strong candidate. M proteins are major virulence factors of group A β-hemolytic Streptococci, and more than 80 serotypes are known, reflecting significant variability in protein sequence thought to assist in warding off host immune responses.237 The autoimmune disorder rheumatic fever is only associated with a few M protein serotypes. In contrast, no specific serotype is associated with guttate psoriasis. All M proteins have conserved amino acids with significant homology to keratins, particularly keratins K16 and K17.238–240 These keratins are strongly upregulated in psoriasis lesions but are either not expressed in normal skin or only at low levels at sites of predilection (i.e., elbows, knees, scalp).241 During the transition from guttate to chronic plaque psoriasis, T cells may recognize the amino acid sequences shared between M proteins, K16, and K17.230 In support of this hypothesis, CD8+ T cells taken from HLA-Cw6-positive patients respond to peptide sequences common to K17 and M protein, whereas nonpsoriatic HLA-Cw6-positive controls only respond to M protein peptides.242 Notably, responsive cells were enriched tenfold in the skin-homing (CLA+) T-cell subset.242 Whereas all HLA-C alleles efficiently present peptides derived from streptococcal M proteins to CD8+ skin-homing memory T cells, HLA-Cw6 may be particularly efficient at presenting M protein-like peptides derived from K16 and K17.
If psoriasis is mediated by antigen-specific T cells, then what are the T cells reacting against? In guttate psoriasis, streptococcal M protein is a strong candidate. M proteins are major virulence factors of group A β-hemolytic Streptococci, and more than 80 serotypes are known, reflecting significant variability in protein sequence thought to assist in warding off host immune responses.237 The autoimmune disorder rheumatic fever is only associated with a few M protein serotypes. In contrast, no specific serotype is associated with guttate psoriasis. All M proteins have conserved amino acids with significant homology to keratins, particularly keratins K16 and K17.238–240 These keratins are strongly upregulated in psoriasis lesions but are either not expressed in normal skin or only at low levels at sites of predilection (i.e., elbows, knees, scalp).241 During the transition from guttate to chronic plaque psoriasis, T cells may recognize the amino acid sequences shared between M proteins, K16, and K17.230 In support of this hypothesis, CD8+ T cells taken from HLA-Cw6-positive patients respond to peptide sequences common to K17 and M protein, whereas nonpsoriatic HLA-Cw6-positive controls only respond to M protein peptides.242 Notably, responsive cells were enriched tenfold in the skin-homing (CLA+) T-cell subset.242 Whereas all HLA-C alleles efficiently present peptides derived from streptococcal M proteins to CD8+ skin-homing memory T cells, HLA-Cw6 may be particularly efficient at presenting M protein-like peptides derived from K16 and K17.
 Immunological tolerance must be overcome for the transition from guttate cross-reactivity to chronic plaque autoreactivity to occur (see Chapter 10). Breakage of tolerance requires high expression of self-antigen,243 and certainly keratins K16 and K17 fulfill this requirement because they are markedly overexpressed in the context of regenerative maturation.241,244 However, many other proteins are markedly upregulated in psoriatic lesions compared to normal skin,245–251 and thus are also candidates for loss of tolerance. Interestingly, many of the most strongly upregulated genes in psoriasis are located in the epidermal differentiation complex (PSORS4, 1q21.3), where genetic linkage and association to psoriasis has been reported.42,43,252 As noted earlier, T-reg function is markedly decreased in psoriasis.111 T-reg dysfunction may further lower the threshold for autoreactivity, as depletion of T-regs is associated with at least tenfold expansion253 and activity254 of the CD8+ T-cell population. With tolerance broken, one would expect that those T cells displaying the highest affinity for self-peptides in the context of HLA-Cw6 would be preferentially stimulated and proliferate, leading to the clonal expansion of CD8+ T cells that is observed experimentally.88
Immunological tolerance must be overcome for the transition from guttate cross-reactivity to chronic plaque autoreactivity to occur (see Chapter 10). Breakage of tolerance requires high expression of self-antigen,243 and certainly keratins K16 and K17 fulfill this requirement because they are markedly overexpressed in the context of regenerative maturation.241,244 However, many other proteins are markedly upregulated in psoriatic lesions compared to normal skin,245–251 and thus are also candidates for loss of tolerance. Interestingly, many of the most strongly upregulated genes in psoriasis are located in the epidermal differentiation complex (PSORS4, 1q21.3), where genetic linkage and association to psoriasis has been reported.42,43,252 As noted earlier, T-reg function is markedly decreased in psoriasis.111 T-reg dysfunction may further lower the threshold for autoreactivity, as depletion of T-regs is associated with at least tenfold expansion253 and activity254 of the CD8+ T-cell population. With tolerance broken, one would expect that those T cells displaying the highest affinity for self-peptides in the context of HLA-Cw6 would be preferentially stimulated and proliferate, leading to the clonal expansion of CD8+ T cells that is observed experimentally.88
 Interestingly, HLA-Cw6 homozygotes have a 2.5-fold higher risk of psoriasis relative to heterozygotes, without having more severe disease.255 This would be the expected outcome if the density of HLA-Cw6 molecules on the surface of DCs together with the local concentration of self-antigen determined the probability of exceeding the threshold for breakage of tolerance, leading to an “all-or-none” process of T-cell activation.230 On the other hand, about half of psoriasis patients are HLA-Cw6 negative, more so in late-onset psoriasis patients lacking a positive family history.256 A recent study indicates that similar mechanisms of antigen recognition and subsequent clonal T-cell expansion are involved in HLA-Cw6-negative patients, though the process may be somewhat more efficient in HLA-Cw6-positive patients.257
Interestingly, HLA-Cw6 homozygotes have a 2.5-fold higher risk of psoriasis relative to heterozygotes, without having more severe disease.255 This would be the expected outcome if the density of HLA-Cw6 molecules on the surface of DCs together with the local concentration of self-antigen determined the probability of exceeding the threshold for breakage of tolerance, leading to an “all-or-none” process of T-cell activation.230 On the other hand, about half of psoriasis patients are HLA-Cw6 negative, more so in late-onset psoriasis patients lacking a positive family history.256 A recent study indicates that similar mechanisms of antigen recognition and subsequent clonal T-cell expansion are involved in HLA-Cw6-negative patients, though the process may be somewhat more efficient in HLA-Cw6-positive patients.257
 How Antigen-Specific Is T-Cell Activation in Psoriasis?
How Antigen-Specific Is T-Cell Activation in Psoriasis? Streptococcally induced guttate psoriasis lacks clonal T-cell receptor gene rearrangements,258 and clonal T-cell receptor rearrangements are not invariably seen in early psoriatic lesions.259 Indeed, most dermal and epidermal T cells found in psoriatic lesions are not clonally expanded. Thus, these T cells may not be directly recognizing antigens at all, instead being “bystanders” supported by the local cytokine environment. Alternatively, it is also possible that many different cross-reactive T-cell clones may recognize self-peptides in the context of HLA-Cw6 with low affinity, without provoking marked expansion. Innate immune mechanisms may also serve to polyclonally activate existing skin-homing memory T cells in psoriasis, especially during the presumed initial infection-related triggering event. Based on the observation of peptidoglycan (PG)-containing macrophages in the papillary and perivascular infiltrates of guttate and chronic plaque psoriasis, it has been suggested that PG, a major constituent of the streptococcal cell wall, may function to activate T cells in psoriasis via a Toll-like receptor (TLR)-mediated and cytokine-dependent mechanism.260 Many PG-reactive T-cell clones have been recovered from psoriasis lesions, suggesting that PG may also serve as a true antigen.260
Streptococcally induced guttate psoriasis lacks clonal T-cell receptor gene rearrangements,258 and clonal T-cell receptor rearrangements are not invariably seen in early psoriatic lesions.259 Indeed, most dermal and epidermal T cells found in psoriatic lesions are not clonally expanded. Thus, these T cells may not be directly recognizing antigens at all, instead being “bystanders” supported by the local cytokine environment. Alternatively, it is also possible that many different cross-reactive T-cell clones may recognize self-peptides in the context of HLA-Cw6 with low affinity, without provoking marked expansion. Innate immune mechanisms may also serve to polyclonally activate existing skin-homing memory T cells in psoriasis, especially during the presumed initial infection-related triggering event. Based on the observation of peptidoglycan (PG)-containing macrophages in the papillary and perivascular infiltrates of guttate and chronic plaque psoriasis, it has been suggested that PG, a major constituent of the streptococcal cell wall, may function to activate T cells in psoriasis via a Toll-like receptor (TLR)-mediated and cytokine-dependent mechanism.260 Many PG-reactive T-cell clones have been recovered from psoriasis lesions, suggesting that PG may also serve as a true antigen.260
 Cross-Priming in Psoriasis: Why Is It Important?
Cross-Priming in Psoriasis: Why Is It Important? Along with K16 and K17, most of the proteins that are strongly overexpressed in psoriatic epidermis are intracellular proteins expressed by keratinocytes. However, keratinocytes are not effective APCs for naive or even memory T cells.261 How, then, could these proteins be effectively processed and presented to CD8+ T cells to generate persistent autoreactivity? Antigen-presenting DCs ingest intracellular proteins from other cells and present them in the context of HLA Class I on their cell surfaces.262 Known as cross-presentation, this process is important in host defense because CD8+ T cells, which are required to kill virus-infected or cancerous cells, only recognize antigen in the context of HLA Class I, and, in epithelial cells, Class I molecules can only be loaded with peptides derived from the intracellular milieu. Many viruses have evolved to exploit this weakness by “learning” not to infect DCs. However, this mechanism carries a risk of autoimmunity, as normal self-proteins are constantly being cross-presented. To minimize this risk, successful activation of a cognate CD8+ T cell requires further support by activated CD4+ T cells, which must also be specific for the infected/cancerous target cell. This process is termed “cross-priming.”263 DCs are the only APCs able to cross-prime CD8+ T cells.264 CD4+ T-cell support is also necessary for maintaining CD8+ effector T-cell function after cross-priming takes place.265,266 In the transition from cross-reactivity to autoreactivity, we envision that CD4+ T cells support the cross-priming of CD8+ T cells capable of recognizing K16/K17 or other intracellularly derived peptides in the context of HLA-Cw6 (Fig. 18-3). Cross-priming would explain the requirement for CD4+ cells during lesional development observed in a xenograft model.267
Along with K16 and K17, most of the proteins that are strongly overexpressed in psoriatic epidermis are intracellular proteins expressed by keratinocytes. However, keratinocytes are not effective APCs for naive or even memory T cells.261 How, then, could these proteins be effectively processed and presented to CD8+ T cells to generate persistent autoreactivity? Antigen-presenting DCs ingest intracellular proteins from other cells and present them in the context of HLA Class I on their cell surfaces.262 Known as cross-presentation, this process is important in host defense because CD8+ T cells, which are required to kill virus-infected or cancerous cells, only recognize antigen in the context of HLA Class I, and, in epithelial cells, Class I molecules can only be loaded with peptides derived from the intracellular milieu. Many viruses have evolved to exploit this weakness by “learning” not to infect DCs. However, this mechanism carries a risk of autoimmunity, as normal self-proteins are constantly being cross-presented. To minimize this risk, successful activation of a cognate CD8+ T cell requires further support by activated CD4+ T cells, which must also be specific for the infected/cancerous target cell. This process is termed “cross-priming.”263 DCs are the only APCs able to cross-prime CD8+ T cells.264 CD4+ T-cell support is also necessary for maintaining CD8+ effector T-cell function after cross-priming takes place.265,266 In the transition from cross-reactivity to autoreactivity, we envision that CD4+ T cells support the cross-priming of CD8+ T cells capable of recognizing K16/K17 or other intracellularly derived peptides in the context of HLA-Cw6 (Fig. 18-3). Cross-priming would explain the requirement for CD4+ cells during lesional development observed in a xenograft model.267
(Fig. 18-5). As reviewed earlier, in the online edition, the advent of GWAS has identified an increasing number of convincing genetic association signals for psoriasis outside the MHC (Table 18-1). The genes contained within these associated regions fit very well with our current concepts of psoriasis pathogenesis. This integration is further reinforced by the pronounced clinical responsiveness of psoriasis to biological agents that specifically target the genetically implicated pathways. Most of the non-MHC associations identified thus far fall into four interconnected functional axes: (1) IFN-γ/IL-23/IL-17 signaling, (2) NF-κB signaling, (3) inflammatory DC function, and (4) keratinocyte differentiation. While the actual functional genetic variations that are ultimately responsible for these associations remain to be determined, these discoveries provide a rationale for biological and therapeutic dissection of the implicated pathways.
Figure 18-5

Proposed model integrating the genetics and immunology of psoriasis. The majority of the CD8+ T-cells (lilac) are located in the epidermis, whereas CD4+ T-cells (blue) predominate in the dermis along with antigen presenting cells/dendritic cells (DCs) (blue) and macrophages (Mφs)(light blue). Confirmed association signals are indicated by the likely candidate genes they contain. Please see text for additional details. (Adapted from Nair RP et al: Psoriasis bench to bedside: Genetics meets immunology. Arch Dermatol 145(4):462-464, 2009, with permission.)
Three strong regions of association map near genes involved in IL-23 signaling: (1) IL12B (encoding the p40 subunit of IL-23 and IL-12), (2) IL23A (encoding the p19 subunit of IL-23), and (3) IL23R (encoding a subunit of the IL-23 receptor).34,60,61 These associations are further supported by the impressive efficacy of biologics targeting the p40 subunit common to IL-12 and IL-23,268 along with the fact that IL12B and IL23A are markedly overexpressed in psoriatic lesions, whereas IL12A is not.269 IL-23 signaling promotes cellular immune responses by promoting the survival and expansion of a subset of IL-17-expressing T cells that protects epithelia against microbial pathogens.270 Given the similarities between skin and gut as epithelial tissues, it is notable that inflammatory bowel disease (IBD) is clinically associated with psoriasis271 and the same genetic variation in IL23R that increases risk for both diseases.272 Ankylosing spondylitis (AS) is another HLA-Class I-associated autoimmune disorder that is clinically associated with IBD273 and genetically associated with IL23R.274 Psoriatic arthritis (PsA) shares a number of clinical similarities with AS, and is genetically associated with IL12B, IL23A, and IL23R34,275,276 (see Chapter 19).
IFN-γ is a major product of activated Th1 cells that stimulates DC to produce IL-23.190 Several psoriasis susceptibility loci contain genes involved in interferon signaling (IFIH1, IL28R, and TYK2)61 Together with IL-1, IL-23 promotes the survival and proliferation of IL-17-expressing T cells (Th17), thereby explaining why Th1 and Th17 are colocalized in psoriasis lesions and in many other sites of inflammation.190 Given the well-known reciprocal relationship between Th1 and Th2 differentiation, it is notable that another psoriasis susceptibility region contains the IL4 and IL13 genes.34,63 In addition to biasing T-cell differentiation away from Th1 and toward Th2, IL-4 inhibits Th17 cell development.277 Both IL-4 and IL-13 are expressed at high levels in atopic dermatitis, but only at very low levels in psoriasis.278 Moreover, treatment of psoriasis with IL-4 resulted in significant clinical improvement,279 accompanied by reduced expression of IL-23 (but not of IL-12) and reduced numbers of Th17 cells.280 The fact that significant genetic signals at both ends of this polarizing spectrum (IL-23, on the one hand, and IL-4/IL-13, on the other), with IFN-γ positioned between them, suggests that Th1-Th2-Th17 balance is a key functional and genetic determinant of psoriasis.
At least five psoriasis-associated genomic regions contain genes involved with controlling signaling through the transcription factor NF-κB: (1) TNFAIP3, (2) TNIP1, (3) NFKBIA, (4) FBXL19, and (5) TRAF3IP2.34,58,59,61,64 TNF-α is a major activator of NF-κB signaling, and these associations are clinically reinforced by the dramatic therapeutic response of psoriasis to anti-TNF biologicals (see Section “Treatment”). TNFAIP3 and TNIP1, respectively encode A20 and ABIN-1, which interact with each other to regulate the ubiquitin-mediated destruction of IKKγ/NEMO, a central nexus of NF-κB signaling.281 TNFAIP3 is genetically associated with rheumatoid arthritis (RA),282,283 and both TNFAIP3 and TNIP1 are associated with systemic lupus erythematosus (SLE).284–287 The polymorphisms implicated in RA and SLE show no association with psoriasis, suggesting that each of these common autoimmune diseases is driven by a different variant of the TNFAIP3 gene. Interestingly, in mice, Tnfaip3 is associated with atherosclerosis,288 which is now known to be a major comorbidity of psoriasis.289 NFKBIA encodes IκBα, which inhibits NF-κB signaling by sequestering it in the cytoplasm. FBXL19 and TRAF3IP2 are significantly more strongly associated with PsA than with purely cutaneous psoriasis.58,64 FBXL19 is structurally related to FBXL11, an F-box family member recently shown to inhibit NF-κB activity by lysine demethylation.290 FBXL11 contains domains known to be required for demethylase activity, but FBLX19 does not.291 Thus, FBXL19 might act as a dominant negative inhibitor of demethylase activity, thereby serving to activate NF-κB. TRAF3IP2 encodes Act1, a ubiquitin ligase that interacts with TRAF (tumor necrosis factor receptor-associated factors) proteins and the IKK complex to activate NF-κB. Interestingly, Act1 is a key component of IL-17-mediated signaling through the IL-17 receptor, allowing the incorporation of TRAF6 into the IL-17 receptor signaling complex, with consequent activation of NF-κB.292 Thus, Act1 may serve as a key link between the IFN-γ/IL-23/IL-17 axis on the one hand, and the NF-κB axis on the other.
Beyond the major role of HLA-Cw6 described earlier, it is noteworthy that two other regions of association contain genes whose products function in antigen presentation: (1) PSMA6, which encodes a proteosomal subunit involved in MHC Class I antigen processing,64 and (2) ERAP1, an IFN-γ-inducible aminopeptidase that trims peptides for optimal binding to the MHC Class I peptide groove. As noted above, inflammatory DC produce large amounts of TNF-α and nitric oxide in addition to their well-recognized functions in antigen presentation. Thus, it seems more than coincidental that another novel region of association contains NOS2, which encodes iNOS, the enzyme responsible for nitric oxide production by DC. Reflective of the massive increase in inflammatory dermal dendritic cells characteristic of psoriasis lesions, NOS2 is markedly overexpressed in lesional skin.64 In addition to iNOS, these inflammatory DC produce large amounts of TNF-α, which synergizes strongly with IL-17 to induce a marked increase expression of innate inflammatory mediators such as hBD2 by keratinocytes.58
Given that hyperproliferation and altered differentiation of keratinocytes figure prominently in psoriasis pathophysiology, it is perhaps surprising that relatively few of the psoriasis-associated regions highlight genes that function primarily in keratinocytes. The most well-established association is an insertion–deletion polymorphism of the late cornified envelope genes LCE3B and LCE3C, which was independently discovered in European65 and Chinese57 populations. Located in the EDC, these genes are expressed very late during keratinocyte terminal differentiation293 and are markedly overexpressed in psoriasis,294 wound healing, and epidermal stress.65,293 Another reported genetic association involving keratinocytes involves increased DEFB4 copy number.66 Whether or not this association is ultimately confirmed, it is notable that DEFB4 is one of the most highly overexpressed genes in psoriatic lesions.295 Finally, TRAF3IP2 is known to function in epidermal cells, as shRNA-mediated knockdown of TRAF3IP2 abrogates the TNF-α + IL-17-mediated upregulation of DEFB4 in human keratinocytes.58
Clinical Findings
Figure 18-6

Diagnosis and treatment algorithm for psoriasis. The diagnosis of psoriasis is usually based on clinical features. In those few cases in which clinical history and examination is not diagnostic, biopsy is indicated to establish the correct diagnosis. The majority of psoriasis cases fall into three major categories: guttate, erythrodermic/pustular, and chronic plaque, of which the latter is by far the most common. Guttate psoriasis is often a self-limited disease with spontaneous resolution within 6–12 weeks. In mild cases of guttate psoriasis, treatment may not be needed, but, with widespread disease, ultraviolet B (UVB) phototherapy or narrowband UVB in association with topical therapy is very effective. Erythrodermic/pustular psoriasis is often associated with systemic symptoms and necessitates treatment with fast-acting systemic medications. The most commonly used drug for erythrodermic and pustular psoriasis is acitretin. In occasional cases of pustular psoriasis, systemic steroids may be indicated (**). Dotted arrows indicate that guttate, erythrodermic, and pustular forms often evolve into chronic plaque psoriasis. Therapeutic choices for chronic plaque psoriasis are typically based on the extent of the disease. Among the main treatment regimens (topical treatment, phototherapy, day treatment centers, and systemic treatments), first-line and second-line modalities are indicated by the solid and dashed lines, respectively. Individuals with conditions that limit their activities, including painful palmoplantar involvement and psoriatic arthritis, may require more potent treatments irrespective of the extent of affected body surface area. Likewise, psychological issues and the impact on quality of life should be taken into consideration. Within each treatment regimen, first-line and second-line choices are grouped. Cyclosporin A is not considered a first-line long-term systemic treatment due to its side effects, but short-term treatment can be helpful for induction of remission. If patients have incomplete response to or are unable to tolerate individual first-line systemic medications, combination regimens (Table 18-6), rotational treatments, or use of biologic therapies should be considered. BB-UVB = broadband UVB; BSA = body surface area; DDx = differential diagnosis; FAE = fumaric acid ester; NB-UVB = narrowband UVB; PUVA = psoralen, and UVA light; tx = therapy.
It is useful to determine the age at onset and the presence or absence of a family history of psoriasis, as younger age of onset and positive family history have been associated with more widespread and recurrent disease.6,21 In addition, the physician should inquire about the prior course of the disease, as major differences exist between “acute” and “chronic” disease. In the latter form, lesions may persist unchanged for months or even years, whereas acute disease shows sudden outbreak of lesions within a short time (days). Likewise, patients have great variability in regard to relapses. Some patients have frequent relapses occurring weekly or monthly, whereas others have more stable disease with only occasional recurrence. The frequently relapsing patients tend to develop more severe disease with rapidly enlarging lesions covering significant portions of the body surface296 and may require more rigorous treatment compared to those with more stable disease. Certain medications may worsen psoriasis (see Section “Treatment”). The physician should also inquire about joint complaints. Although osteoarthritis is extremely common and can coexist with psoriasis, a history of onset of joint symptoms before the fourth decade and/or a history of warm, swollen joints should raise the suspicion of psoriatic arthritis (see Chapter 19).
The classic lesion of psoriasis is a well-demarcated, raised, red plaque with a white scaly surface (Fig. 18-7). Lesions can vary in size from pinpoint papules to plaques that cover large areas of the body. Under the scale, the skin has a glossy homogeneous erythema, and bleeding points appear when the scale is removed, traumatizing the dilated capillaries below (the Auspitz sign) (Fig. 18-8).297 Psoriasis tends to be a symmetric eruption, and symmetry is a helpful feature in establishing a diagnosis. Unilateral involvement can occur, however. The psoriatic phenotype may present a changing spectrum of disease expression even within the same patient.


Koebner phenomenon (also known as the isomorphic response) is the traumatic induction of psoriasis on nonlesional skin; it occurs more frequently during flares of disease and is an all-or-none phenomenon (i.e., if psoriasis occurs at one site of injury it will occur at all sites of injury) (Fig. 18-9). The Koebner reaction usually occurs 7–14 days after injury, and approximately 25% of patients may have a history of trauma-related Koebner phenomenon at some point in their lives.298 Estimates of lifetime prevalence rise as high as 76% when factors such as infection, emotional stress, and drug reactions are included.4 The Koebner phenomenon is not specific for psoriasis but can be helpful in making the diagnosis when present.

Psoriasis vulgaris is the most common form of psoriasis, seen in approximately 90% of patients. Red, scaly, symmetrically distributed plaques are characteristically localized to the extensor aspects of the extremities, particularly the elbows and knees, along with scalp, lower lumbosacral, buttocks, and genital involvement (see Fig. 18-7). Other sites of predilection include the umbilicus and the intergluteal cleft. The extent of involvement varies widely from patient to patient. There is constant production of large amounts of scale with little alteration in shape or distribution of individual plaques. Single small lesions may become confluent, forming plaques in which the borders resemble a land map (psoriasis geographica). Lesions may extend laterally and become circinate because of the confluence of several plaques (psoriasis gyrata). Occasionally, there is partial central clearing, resulting in ring-like lesions (annular psoriasis) (Fig. 18-10). This is usually associated with lesional clearing and portends a good prognosis. Other clinical variants of plaque psoriasis have been described depending on the morphology of the lesions; particularly those associated with gross hyperkeratosis (see Fig. 18-10). Rupioid psoriasis refers to lesions in the shape of a cone or limpet. Ostraceous psoriasis, an infrequently used term, refers to a ring-like, hyperkeratotic concave lesion, resembling an oyster shell. Finally, elephantine psoriasis is an uncommon form characterized by thickly scaling, large plaques, usually on the lower extremities. A hypopigmented ring (Woronoff ring) surrounding individual psoriatic lesions may occasionally be seen and is usually associated with treatment, most commonly UV radiation or topical corticosteroids (see Fig. 18-10C). The pathogenesis is not well understood but may result from inhibition of prostaglandin synthesis.299
Figure 18-10

Unusual forms of plaque-type psoriasis A. Annular psoriasis on the flank. B. Rupioid psoriasis in an infant. Note cone-shaped lesions. C. Psoriatic patient undergoing modified Goeckerman therapy (ultraviolet B light, coal tar, and topical steroid), demonstrating Woronoff rings. D. Elephantine psoriasis of the lower extremities. Note psoriatic involvement of toenails. (Photographs used with permission from Dr. Johann Gudjonsson, Mr. Harrold Carter, and Ms. Laura Vangoor.)
Guttate psoriasis (from the Latin gutta, meaning “a drop”) is characterized by eruption of small (0.5–1.5 cm in diameter) papules over the upper trunk and proximal extremities (Fig. 18-11). It typically manifests at an early age and as such is found frequently in young adults. This form of psoriasis has the strongest association to HLA-Cw6,26 and streptococcal throat infection frequently precedes or is concomitant with the onset or flare of guttate psoriasis.300 However, antibiotic treatment has not been shown to be beneficial or to shorten the disease course.301 Patients with a history of chronic plaque psoriasis may develop guttate lesions, with or without worsening of their chronic plaques.

Small plaque psoriasis resembles guttate psoriasis clinically, but can be distinguished by its onset in older patients, by its chronicity, and by having somewhat larger lesions (typically 1–2 cm) that are thicker and scalier than in guttate disease. It is said to be a common adult-onset presentation of psoriasis in Korea and other Asian countries.141
Psoriasis lesions may be localized in the major skin folds, such as the axillae, the genito-crural region, and the neck. Scaling is usually minimal or absent, and the lesions show a glossy sharply demarcated erythema, which is often localized to areas of skin-to-skin contact (Fig. 18-12). Sweating is impaired in affected areas.302

(See also Chapter 23). Psoriatic erythroderma represents the generalized form of the disease that affects all body sites, including the face, hands, feet, nails, trunk, and extremities (Fig. 18-13). Although all the symptoms of psoriasis are present, erythema is the most prominent feature, and scaling is different compared with chronic stationary psoriasis. Instead of thick, adherent, white scale there is superficial scaling. Patients with erythrodermic psoriasis lose excessive heat because of generalized vasodilatation, and this may cause hypothermia. Patients may shiver in an attempt to raise their body temperature. Psoriatic skin is often hypohidrotic due to occlusion of the sweat ducts,303 and there is an attendant risk of hyperthermia in warm climates. Lower extremity edema is common secondary to vasodilatation and loss of protein from the blood vessels into the tissues. High-output cardiac failure and impaired hepatic and renal function may also occur. Psoriatic erythroderma has a variable presentation, but two forms are thought to exist.304 In the first form, chronic plaque psoriasis may worsen to involve most or all of the skin surface, and patients remain relatively responsive to therapy. In the second form, generalized erythroderma may present suddenly and unexpectedly or result from nontolerated external treatment (e.g., UVB, anthralin), thus representing a generalized Koebner reaction. Generalized pustular psoriasis [see Section “Generalized Pustular Psoriasis (von Zumbusch)”] may revert to erythroderma with diminished or absent pustule formation. Occasional diagnostic problems may arise in differentiating psoriatic erythroderma from other causes (see Chapter 23).
Figure 18-13

Erythrodermic psoriasis. The patient shown in panel A rapidly developed near-complete involvement and complained of fatigue and malaise. Note islands of sparing. The patient shown in panels B and C had total body involvement with marked hyperkeratosis and desquamation. (Photos used with permission from Mr. Harrold Carter and Dr. Johann Gudjonsson.)
Several clinical variants of pustular psoriasis exist: generalized pustular psoriasis (von Zumbusch type), annular pustular psoriasis, impetigo herpetiformis, and two variants of localized pustular psoriasis—(1) pustulosis palmaris et plantaris and (2) acrodermatitis continua of Hallopeau. In children, pustular psoriasis can be complicated by sterile, lytic lesions of bones305,306 and can be a manifestation of the SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, osteitis).307
Generalized pustular psoriasis (von Zumbusch) is a distinctive acute variant of psoriasis. It is usually preceded by other forms of the disease. Attacks are characterized by fever that lasts several days and a sudden generalized eruption of sterile pustules 2–3 mm in diameter (Fig. 18-14). The pustules are disseminated over the trunk and extremities, including the nail beds, palms, and soles. The pustules usually arise on highly erythematous skin, first as patches (Fig. 18-14C) and then becoming confluent as the disease becomes more severe. With prolonged disease, the fingertips may become atrophic. The erythema that surrounds the pustules often spreads and becomes confluent, leading to erythroderma. Characteristically, the disease occurs in waves of fevers and pustules. The etiology of generalized psoriasis von Zumbusch type is unknown. Various provoking agents include infections, irritating topical treatment (Koebner phenomenon), and withdrawal of oral corticosteroids.308 This form of psoriasis is usually associated with prominent systemic signs and can potentially have life-threatening complications such as hypocalcemia,309 bacterial superinfection, sepsis, and dehydration.310 Severe pustular psoriasis can be difficult to control and requires a potent treatment regimen with rapid onset of action to avoid life-threatening complications. Drugs commonly used include etretinate, methotrexate (MTX), cyclosporine, infliximab,311,312 or oral corticosteroids (see Section “Treatment”).313 Cases of acute respiratory distress syndrome associated with generalized pustular psoriasis have been reported.314 Two recent reports have identified recessive loss-of function mutations in the IL36RN gene encoding IL-36 receptor antagonist (IL-36ra) in patients with generalized pustular psoriasis. Consistent with the prominent inflammation associated with generalized pustular psoriasis, IL-36ra is an anti-inflammatory cytokine that inhibits signaling by three related IL-1 like proteins (IL-36alpha, beta, and gamma).315,316
Figure 18-14

Pustular psoriasis. A and B, von Zumbusch-type generalized pustular psoriasis. Note tiny pustules, 1–2 mm in diameter, on erythematous skin. C and D, localized pustular psoriasis of the leg and foot, respectively. E, resolving pustular psoriasis, note extensive areas of desquamation. (Photos C–E used with permission from Drs. Johann Gudjonsson, Trilokraj Tejasvi, and Neena Khanna.)
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


