Disorders of Hair and Nails
David Whiting M.D.
Bernice Krafchik M.D.
Richard Scher M.D.
Kurt Hirschhorn M.D.
Judith Willner M.D.
Clinical Pearls
(DW)
(BK)
(RS)
(KH)
(JW)
Menkes’ Syndrome
Synonym
Menkes kinky hair syndrome
Occipital horn syndrome (OHS)
Inheritance
X-linked recessive; MKN or ATP7A gene on Xq13
Prenatal Diagnosis
DNA analysis
Amniocentesis/chorionic villus sampling (CVS)—increased incorporation of copper by cultured amniotic fluid cells
Incidence
1:35,000 in Australia, 1:300,000 in Europe; males only; approximately 90% with classic, severe form, 10% with milder OHS
Age at Presentation
First few months of life
Pathogenesis
Mutations in MKN or ATP7A, a gene encoding the copper-binding enzyme adenosine triphosphatase (ATPase), leads to defective copper transport and metabolism with subsequent low levels of serum copper; phenotype reflects deficiency of copper-dependent enzyme activity in various systems
Key Features
Hair
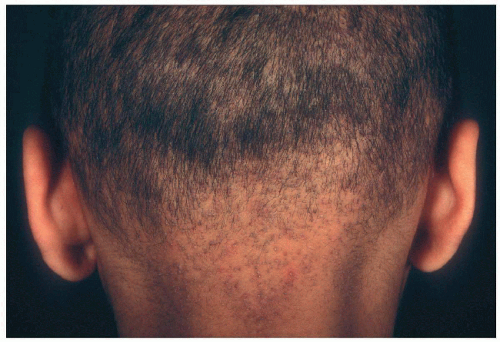
Pili torti most common; trichorrhexis nodosa, monilethrix described; hypopigmented, sparse, short, brittle, “steel-wool” quality; sparse, broken horizontal eyebrows; sparse eyelashes
Skin
Hypopigmented, “doughy” consistency with laxity, pudgy cheeks, Cupid’s bow upper lip
Central Nervous System
Progressive deterioration with lethargy, seizures, mental and motor retardation, hypertonia, hypothermia
Musculoskeletal
Failure to thrive, frontal bossing, wormian bones in sagittal and lambdoid sutures, metaphyseal widening with spurs in long bones, fractures
OHS: occipital horns (exostosis at insertion of trapezius and sternocleidomastoid muscles), abnormal facies, short flat clavicles, elbow deformities secondary to radial subluxation
Cardiovascular
Tortuous arteries (especially brain)
Genitourinary
Variety of anomalies
Differential Diagnosis
Battered child syndrome
Argininosuccinic aciduria (p. 278)
Björnstad syndrome (p. 276)
Laboratory Data
Serum copper, ceruloplasmin levels DNA analysis
Management
Parenteral copper histidine if initiated first 8 weeks of life may be of benefit Antiseizure medications, pamidronate has been helpful in preventing fractures in one study
Prognosis
Progressive deterioration with death by 2-3 years of age associated with pneumonia
Clinical Pearls
Can have an awful lot of trichorrhexis nodosa as well… They live only about a year or two… Female carriers can have pili torti… Genetic counseling should be stressed. DW
|

 10.1. Three-month-old with doughy, lax skin and “pudgy” face, sparse hair. (104) |
 10.2. Same patient with metaphysical widening of femur and tibia and femoral spurs. Note osteoporosis. (104) |
 10.3. Doughy redundant skin on palm. (104) |
 10.4. “Steel-wool” hair (105) |
Björnstad Syndrome
Inheritance
Autosomal recessive; 2q34-q36 gene locus
Prenatal Diagnosis
None
Incidence
Very rare—approximately 25 cases reported; M=F
Age at Presentation
By 2 years old
Pathogenesis
Unknown
Key Features
Hair
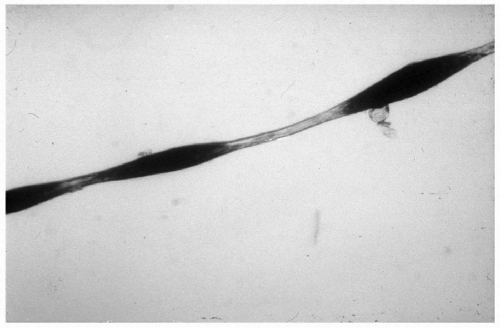
Pili torti with/without alopecia of scalp; eyebrows, eyelashes unaffected
Ear-Nose-Throat
Bilateral sensorineural deafness
Differential Diagnosis
Crandall syndrome (pili torti, deafness, hypogonadism) Menkes’ syndrome (p. 274)
Laboratory Data
Auditory testing
Management
Referral to audiologist
Prognosis
Normal intelligence, life span; hearing loss mild to severe with increased severity associated with more severe hair defects
Clinical Pearls
If you diagnose pili torti in a child, send them all to an audiologist early on to prevent potential speech deficits… They get “classic” pili torti as a rule. DW
|

 10.5. Short, sparse hair in 9-year-old girl. (106) |
 10.6. Pili torti-typical twisting of hair shaft. (106) |
Argininosuccinic Aciduria
Inheritance
Autosomal recessive; argininosuccinate lyase (ASL) gene on 7cen-q11.2
Prenatal Diagnosis
Amniocentesis—argininosuccinase assay in cultured amniotic fluid cells DNA analysis
Incidence
1:70,000 U.S. births; M=F
Age at Presentation
Neonate (neonatal form) or second year of life (late-onset form)
Pathogenesis
Mutation in ASL leads to a deficiency in argininosuccinate lyase—second most common urea cycle defect
Mechanism of hair defect unknown
Key Features
Hair
Trichorrhexis nodosa (approximately 50% affected)—increased with late-onset disease; dull, dry, matted and fragile; increased in occipital region
Musculoskeletal
Failure to thrive (neonatal)
Hematologic
Hyperammonemia
Gastrointestinal
Vomiting
Hepatomegaly (neonatal)
Central Nervous System
Seizures
Lethargy, coma (neonatal)
Ataxia, severe mental retardation (late-onset)
Differential Diagnosis
Citrullinemia
Familial trichorrhexis nodosa
Acquired trichorrhexis nodosa
Laboratory Data
Enzyme assay—argininosuccinase deficiency in red blood cells and cultured fibroblasts
High-voltage electrophoresis or ion-exchange chromatography—increased blood, urine, or cerebrospinal fluid (CSF) argininosuccinic acid levels
Increased ammonia levels in blood
Management
Restricted protein diet (1.0 to 1.5 g/kg per day) with arginine supplementation (3 to 5 mmol/kg per day)
Referral to dermatologist, neurologist
Liver transplant
Prognosis
If survival beyond the neonatal period, most will be mentally retarded; late-onset cases are severely mentally retarded
Clinical Pearls
One child we took care of actually received a liver transplant in Pittsburgh, which is the only available therapy… Unfortunately, she died of fungal sepsis 1 year later… Prenatal linkage studies could not be done on the family… An enormous rarity. KH, JW
Trichorrhexis nodosa is caused by trauma and is often seen in metabolic disorders with brittle hair. DW
|

 10.7. Short, broken scalp hairs. (107) |
 10.8. Scanning-electron micrograph depicting trichorrhexis nodosa. (108) |
Monilethrix
Inheritance
Autosomal dominant; human basic type II hair keratin genes, hHb1 and hHb6 on 12q13
Prenatal Diagnosis
None
Incidence
Rare; M=F
Age of Presentation
First few months of life
Pathogenesis
Mutations in human hair keratin genes expressed in cortical trichocytes of the hair shaft, leads to defect in many cases
Key Features
Hair
Structural defect: elliptical nodes along shaft (0.7 to 1.0 mm apart) with undulating variation in diameter; “beaded” appearance under the light microscope; breaks at internodes
Dry, brittle, lusterless, sparse, short
Scalp most common, but may occur on eyelashes, eyebrows, and body
Skin (most common association)
Keratosis pilaris on upper back, nape of neck, arms
Nails
Brittle
Eyes
Cataracts (rare)
Mouth
Teeth abnormalities
Central Nervous System
Mental retardation (rare)
Differential Diagnosis
None
Laboratory Data
Light microscopy of hair shaft
Management
Referral to dermatologist—avoid hair trauma, retinoids, topical minoxidil
Referral to symptom-specific specialist
Prognosis
May improve with pregnancy and at puberty
Clinical Pearls
You can try isotretinoin or acitretin… I saw a family at a meeting in San Antonio a few years ago where the sons had severe alopecia, while the father had mild alopecia… His had been severe as a boy… Associations not that common… Occasional case reports. DW
|

 10.9. Short, sparse hair with keratosis pilaris on nape of neck. (109) |
 10.10. Monilethrix- “beaded” appearance under light microscopy (110) |
Uncombable Hair Syndrome
Synonym
Pili trianguli et canaliculi
Spun-glass hair
Inheritance
Autosomal dominant in some cases; gene locus unknown
Prenatal Diagnosis
None
Incidence
Approximately 50 cases reported; M=F
Age at Presentation
Usually in infancy
Pathogenesis
Unknown
Key Features
Hair
Blonde, dry, shiny hair—unable to comb into place, not fragile
Eyelashes, eyebrows unaffected
Differential Diagnosis
None
Laboratory Data
Electron microscopy—canal-like groove along shaft of a triangular-shaped hair
Management
Biotin 0.3 mg three times per day may help
Prognosis
May improve with age
Clinical Pearls
Doesn’t always improve with age, but it may… Not fragile hair… Just difficult to comb… Walter Shelley has found that biotin helped to manage the hair in one case… He sent me hair from the case, and I found the structural defect unchanged… Looking under the light microscope, I had no problem finding longitudinal grooves… No common associations. DW
|

 10.11. Shiny, “spun glass” hair. Note normal eyebrows, eyelashes. (111) |
 10.12. Close-up of scalp hair. (111) |
Hypohidrotic Ectodermal Dysplasia
Synonym
Anhidrotic ectodermal dysplasia
Christ-Siemens-Touraine syndrome
Inheritance
X-linked recessive—ectodysplasin (EDA) gene on Xq 12-q 13
Autosomal dominant, recessive and other x-linked (NEMO mutations) cases described but rare
Prenatal Diagnosis
DNA analysis
Fetoscopy (20 weeks)—skin biopsy with absent pilosebaceous units
Incidence
Approximately 1:100,000; >90% males; female carriers partially affected
Age at Presentation
Infancy to early childhood
Pathogenesis
Mutation in ectodysplasin, a member of the tumor necrosis family, leads to defective regulation of ectodermal structures
Key Features
Skin
Smooth, soft, dry, fine wrinkles with pigmentation periorbitally; hypoanhidrosis with hyperpyrexia; increased frequency of atopic dermatitis
Newborn: may have collodion membrane, marked scaling
Hair
Hypopigmented, fine, short, sparse scalp and body hair; longitudinal groove on electron microscopy; eyebrows, eyelashes fine to absent
Nails
Slight dystrophy (much less common and insignificant compared to hidrotic ectodermal dysplasia)
Craniofacial
Frontal bossing, saddle nose, prominent supraorbital ridges, everted thick lips, hypoplastic midface, abnormal ears
Teeth
Hypo-anodontia, peg-shaped/conical incisors and canines; molars with hooked cusps; deciduous and permanent affected; hypoplastic gum ridges noted early on
Sinopulmonary (less common)
Atrophic rhinitis with thick, foul-smelling discharge; increased bronchopulmonary infection, asthma
Differential Diagnosis
Other ectodermal dysplasias
Laboratory Data
Skin biopsy of palmar skin—lack of eccrine units Jaw film
DNA analysis
Management
Avoid overheating with limits on physical activity, exercise, “cool suit,” appropriate occupation, air conditioning, cool baths, avoid warm climates; close monitoring for infection with early antibiotic intervention, antipyretics
Methyl cellulose 1% drops for dry mucosa of eyes, nose—avoid antihistamines; skin emolliation
Referral to pediatric dentist—dentures, implants
Referral to plastic surgery—facial cosmesis, wig
Referrral to ear-nose-throat (ENT) specialist—manage recurrent infection, asthma
Examine first-degree relatives
Prognosis
May have stunted development, febrile convulsions; rarely fatal early on with improvement in late childhood; otherwise normal life span
Clinical Pearls
Most importantly, avoid overheating… Air conditioning, cool temperatures, light clothing… I have to write to schools and tell them the kids should be in an airconditioned environment… One infant used to drag himself across the marble floor to keep cool… Kids find out for themselves what their exercise limitations are… Our dentist does beautiful work using plates early on and implants as they get older… Avoid extracting teeth so that the alveolar ridge can be maintained… Early acquaintance with the dentist… Difficult diagnosis in nursery but may be recognized by the typical wrinkles around the eye… Look at mother carefully… Carriers often have dry hair, skin, and missing teeth… Nail changes do not occur… They all start to resemble one another… BK
|

 10.13. Indian male with fine, short, sparse hair, frontal bossing, saddle nose, prominent supraorbital ridge, periorbital pigmentation, and thick everted lips. (5) |
 10.14. Anodontia and conical canines. (112) |
Hidrotic Ectodermal Dysplasia
Synonym
Clouston syndrome
Inheritance
Autosomal dominant; connexin 30 (GJB6) gene on 13q11-12
Prenatal Diagnosis
None
Incidence
Rare; most common in French-Canadian and French population; M=F
Age at Presentation
Birth to neonatal period
Pathogenesis
Mutation in connexin 30 leads to defective ectodermal development and maintenance
Key Features
Skin
Palmoplantar keratoderma with transgradiens
Nails
Dystrophy—thickened, milky white early on, micronychia, hyperconvex, longitudinal striations, discolored, brittle, absent
Paronychial infections with/without nail matrix destruction
Hair
Scalp—normal early on but often becomes thin, wiry, brittle, pale, sparse, or absent after puberty
Body, eyelashes, eyebrows—sparse to absent; secondary conjunctivitis, blepharitis
Musculoskeletal
Tufting of terminal phalanges and thickened skull bones may occur
Differential Diagnosis
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


