Introduction
The word “scar” originates from the Greek word eskhara, which means scab. It is defined in the Merriam-Webster online dictionary (2023) as a mark remaining after injured tissue has healed. Morphologically it is usually devoid of skin markings and may be skin colored, erythematous, hypopigmented, or hyperpigmented. Partial-thickness wounds, which involve epidermis and superficial dermis with preservation of skin appendages, tend to heal without scarring. Full-thickness wounds involve deep dermis and heal with scarring—a process that can be normal or abnormal.
Normal Wound Healing
Partial-thickness wounds only involve the epidermis and superficial dermis and do not involve adnexal structures. Inflammation and granulation tissue formation are minimal, and reepithelialization, which occurs from the edges of the wound as well as from adnexal structures, results in complete and rapid healing with minimal or no scarring. Pigmentary changes may, however, still occur at the wound site. In contrast, full-thickness wounds require clot formation to halt bleeding from larger vessels in the deep dermis. Inflammation and granulation are important stages, and contraction is critical in facilitating reepithelialization by bringing the edges of the wound together. In deep wounds, reepithelialization occurs solely from the epithelial margins due to destruction of adnexal structures and, ultimately, scar tissue is formed to replace the tissue loss.
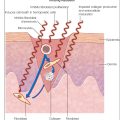
Healing and scar formation is a dynamic process involving three key, and often overlapping, phases: (i) inflammatory phase; (ii) proliferative phase; and (iii) remodeling.
Inflammatory Phase
The inflammatory phase starts within the first 6 to 8 hours of wound formation and can last about 3 to 4 days. It begins with hematoma formation, and the first cells to the site of the wound are platelets. Clot formation arrests hemorrhage from ruptured vessels, provides a temporary barrier to exogenous pathogens, and creates a provisional matrix through which inflammatory cell migration occurs. Sequestered platelets degranulate and release several growth factors including transforming growth factor beta 1 (TGF-β1), epidermal growth factor (EGF), insulin-like growth factor 1 (IGF-1), and platelet-derived growth factor (PDGF). These growth factors initiate signaling pathways leading to recruitment of inflammatory cells, extracellular matrix (ECM) formation, and neovascularization.
A diverse range of other molecules, such as by-products of fibrinolysis, serve as chemotactic signals and recruit neutrophils and monocytes to the wound site. These cells migrate from the circulation to the wound over the first 48 hours of wound formation by diapedesis through the endothelial lining of capillaries adjacent to the wound site. The chief role of neutrophils is phagocytosis and intracellular killing of microorganisms. They also express several proinflammatory cytokines that might serve as early signaling pathways to activate local keratinocytes and macrophages. Additionally, the production of polysaccharide hyaluronan (HA) increases rapidly in the early inflammatory phase and is known to play a key role in all phases of wound healing.
After the acute inflammatory response (24–48 hours), infiltrating monocytes are activated as macrophages and clear residual pathogens and cell and matrix debris. These activated macrophages also serve as another source of growth factors and cytokines such as PDGF, which stimulate the recruitment of fibroblasts into the site of injury. Macrophages are required for proper wound healing and set the stage for the next phase of wound healing.
Proliferative Phase
The proliferative phase of wound healing starts around days 5 to 7 and may last up to 1 month. Early granulation tissue is highly vascular and densely cellular. In full-thickness wounds, which do not heal by reepithelialization alone, dermal fibroblasts adjacent to the wound proliferate, migrate, and produce extracellular matrix (ECM) composed of fibrin, fibronectin, vitronectin, and glycosaminoglycans (GAG). There is a high ratio of collagen III/collagen I in early granulation tissue. Keratinocytes start to proliferate in response to growth factors in the wound. Eventually, keratinocytes from sites adjacent to the wound leapfrog over each other to initiate reepithelialization. Structurally, the lateral mobilization of keratinocytes is due to the secondary breakdown of desmosomes.
As granulation evolves, and an abundant collagen matrix has been deposited, cellularity diminishes due to apoptosis. New blood vessels sprout into the ECM triggered by angiogenesis molecules, inducing vascular endothelial growth factor (VEGF), TGF-β1, angiotropin, and thrombospondin. In large wounds, contraction mediated by transformed wound fibroblasts (myofibroblasts) reduces the quantity of granulation tissue needed to fill the defect and minimizes the area of reepithelialization needed to replace tissue loss. Fibroblasts also provide wound edge tension through contractile proteins actin and desmin. The release of mechanical tension after the wound has closed sends “stop” signals for wound contraction. Keratinocyte migration and reepithelialization is stimulated, and migrating keratinocytes upgrade tissue-type and urokinase-type plasminogen activators (tPA and uPA) and receptors for uPA to activate fibrinolysis, an essential step for keratinocyte migration. In order to mobilize through the provisional matrix, keratinocytes also express receptors for fibronectin and collagen.
Remodeling
During the final stage of wound healing, the remodeling or maturation phase, a balance of matrix degradation and collagen synthesis occurs. Matrix degradation occurs through the removal of excess collagen and ECM by tissue enzymes, such as matrix metalloproteinases, produced by fibroblasts, mast cells, endothelial cells, and macrophages. The presence of inhibitors of these metalloproteinases, such as TIMP I and TIMP II, plays an important role in tissue repair. Fibroblasts synthesize collagen, contractile proteins, and ECM to assist in scar maturation. Interferons produced by T lymphocytes (interferon-gamma), leucocytes (interferon-alpha), and fibroblasts (interferon-beta) have antifibrotic activity and inhibit fibroblast synthesis of collagen and fibronectin.
Scar strength gradually increases over weeks to months. At 1 week, scars are at about 1% tensile strength; at 3 weeks, 20%; at 3 months, 50%; and at 1 year, 80%. Scar tissue is therefore more vulnerable to reinjury. Early scars have a high ratio of collagen III/collagen I. Over the next 6 to 12 months, the wound undergoes significant remodeling, characterized by collagen deposition and cross-linking, resulting in the establishment of a mature scar, mainly replaced by type I collagen. The entire remodeling process occurs over 6 to 12 months but can persist for years after the initial injury.
Abnormal Wound Healing
Cutaneous wounds sometimes heal with exuberant scarring to form hypertrophic scars or keloid scars. The basis for abnormal scarring has not been fully elucidated, but it is known that hypertrophic scars and keloids depart early from the normal wound healing process and are affected by a number of factors on a cellular and molecular level. The following mechanisms have been suggested as playing key roles in pathologic wound healing and abnormal scar formation:
Collagen and Collagenase
Both an increase in amount and duration of collagen synthesis contribute to abnormal wound healing of keloids. Formation of new collagen following the inflammatory stage extends over a much longer period of time than in normal healing, and collagen synthesis is significantly (approximately 20 times) higher than in normal skin, with increased turnover, resulting in a disproportionate amount of collagen deposition. Fibroblasts from keloid scars produce increased amounts of collagen per cell, in vitro as well as in vivo, in the absence of growth factors. Elevated levels of collagenase, as well as collagenase inhibitors, have been found in keloids.
TGF-β
TGF-β has emerged as a likely candidate for inducing hypertrophic scar and keloid formation. TGF-β induces collagen, fibronectin, and proteoglycan synthesis by fibroblasts. It also decreases collagenase production and increases production of collagenase inhibitors, thus reducing matrix degradation. Three isoforms of TGF-β (1, 2, and 3) have been identified in mammals. Keloid fibroblasts are particularly sensitive to isoforms of TGF-β1 and TGF-β2, which have profibrotic functions and increase scar formation. In a controlled study, wound treatment with neutralizing antibodies to TGF-β1 and -β2 reduced collagen deposition and scarring compared to control wounds in rodents. The addition of TGF-β3 reduced the monocyte and macrophage profile, inflammation, fibronectin, and collagen I and III deposition in the early stages of wound healing, improved the architecture of the neodermis, and reduced scarring. It has been reported that TGF-β3 isoforms increased in intensity from day 7 post wounding in old mice, while TGF-β1 and -β2 isoforms were increased at all time points in the wounds of younger mice. This could explain to some degree the improved quality of tissue repair and decreased quantity of scar formation in the older population. The relative proportion of TGF-β isoforms, such as the ratio of TGF-β3 to TGF-β1 may determine the wound repair outcome.
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are tissue enzymes that play a major role in the remodeling of ECM during wound healing. The composition of the ECM in fetal skin and wounds differs from that in adults. It has been suggested that differential expression of MMPs and TIMPs occurs during the ontogenetic transition to scar-forming wound repair. Expression of MMPs and TIMPs appears to be carefully regulated during normal wound healing. An imbalance between MMPs and TIMPs activity can impair wound healing and cause abnormal scar formation. MMP1 is significantly upregulated in both scarless and scarring wounds. However, the maximal increase in expression of MMP1 and MMP9 occurred more rapidly and was much greater in scarless fetal wounds. Although unchanged in scarless wounds, MMP2 levels decreased more than threefold in scarring wounds. Levels of MMP1 and MMP3 were increased in scar-forming wounds. Overall, it appeared that scarless wound repair is associated with a higher ratio of MMP to TIMP expression compared to scarring wounds.
Apoptosis
Apoptosis, a form of programmed cell death, is genetically determined and necessary for maintaining tissue homeostasis. Apoptosis is modulated by several intrinsic and extrinsic signals. In normal wound healing, apoptosis is thought to play a role in the transition from granulation tissue into definitive scar tissue. In this transition period, several cell types, including fibroblasts, inflammatory cells, endothelial cells, and possibly most importantly myofibroblasts, undergo apoptosis. Keloid fibroblasts have been shown to have lower rates of apoptosis than normal skin fibroblasts. Table 1.1 summarizes the proapoptotic/antiapoptotic effects of the genes and proteins that are described in this chapter. Several mechanisms have been suggested as being responsible for this resistance to apoptosis observed in keloidal tissue, which are briefly highlighted below:
- •
p53: The p53 gene is a well-characterized tumor-suppressor gene that functions by controlling cell proliferation and inducing apoptosis; loss-of-function mutations in this gene are associated with a variety of sporadic human tumors. In one study, several p53 gene focal mutations were identified in keloidal fibroblasts, while no mutations were observed in fibroblasts from normal skin of the same patients.
- •
bcl-2: Upregulation of bcl-2, an anti-apoptotic protein, was identified in younger, hypercellular peripheral areas of the keloid and associated with a lower rate of apoptosis compared to controls.
- •
Fas : Expression of Fas, a proapoptotic protein, has been observed to be more prominent in the central, hypocellular regions of the keloids. Fas-mediated apoptosis is preserved in keloidal fibroblasts as compared to hypertrophic scar-derived and normal skin-derived fibroblasts when TGF-β1 was added, which regulates apoptosis in keloid-derived fibroblasts.
- •
Caspases : The caspase family is a group of proapoptotic proteases in which sequential activation leads to a cascade of proteolytic activity and cell death. Caspase-8, -13, and -14 were found to be upregulated in keloids when compared to adjacent normal skin. Caspase-8 is the first enzyme in the cascade activated by the binding of the Fas and TNF receptor. Caspase-13 is also a proapoptotic enzyme that is activated by caspase-8. Interestingly, caspase-14 is not known to participate in a proapoptotic pathway and is only expressed in keratinocytes. Caspase-6, a proapoptotic protein, was found to be downregulated in keloids.
- •
TNF : The TNF superfamily includes both the TNF ligand and the TNF receptor family, which regulates expression of apoptotic machinery. Compared to normal skin, TNFSF12 and TNFSF14 (ligands) were upregulated, whereas TNFRSF10B and TNFRSF1B (receptors) were downregulated. Interestingly, in the presence of TNF-α, fibroblasts from keloids had lower expression of apoptotic genes, primarily from the caspase and TNF superfamily, when compared to normal fibroblasts. This resistance to apoptosis could be explained by the decreased number of TNF receptors on keloidal fibroblasts.
- •
Gene Expression: Sayah and colleagues compared the expression of 64 apoptosis-related genes in keloids and normal scars. Eight of 64 genes were found to be significantly (difference >50%) underexpressed in keloidal tissue: TRADD, c-myc protooncogene, NIP-3, HDLC-1, which are promoters of apoptosis; and DAD-1, G-S-T, G-S-T-M, and glutathione peroxidase, which are apoptosis inhibitors.
| Proapoptotic | Antiapoptotic |
|---|---|
| Caspase 3 | Bcl |
| Caspase 6 | DAD-1 |
| Caspase 8 | ERK |
| c-myc | GST |
| NIP-3 | GSTM |
| HDLC-1 | GP |
| TRADD | JNK |
| TNF a | TNF a |
Epidermal Keratinocytes
Epidermal keratinocytes have been implicated to play a role in dermal fibroblast regulation. Keloidal fibroblasts cultured with keloid-derived keratinocytes proliferated more than those cultured with normal skin keratinocytes. Moreover, keloidal fibroblast cultures with keloidal keratinocytes are more resistant to apoptosis than normal ones cultured with normal skin keratinocytes. Decreased levels of caspase-3 (a proapoptotic factor) and increased expression of bcl-2 and increased ERK and JNK phosphorylation (all antiapoptotic factors) were found in keloidal fibroblasts cultured with keloidal keratinocytes. In addition, Caspase-14, which is expressed only by keratinocytes, is upregulated in keloids, further suggesting a relevant interaction between the epidermis and dermis in keloid biology.
Morphologic and Histologic Features of Abnormal Scars
Hypertrophic Scars
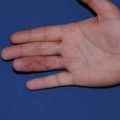
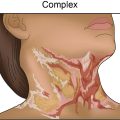
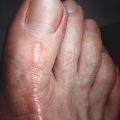
Hypertrophic scars ( Table 1.2 ) are confined to the boundary of the original injury and increase in bulk by pushing out the margin, rather than invasion. They are usually asymptomatic, develop within weeks after the injury, and tend to gradually resolve over time. They are common among younger individuals. They may develop either hypo- or hyperpigmentation and tend to have a smooth surface. Hypertrophic scars ( Figs. 1.1 and 1.2 ) and keloids share some clinical features. For example, the overlying epidermis in hypertrophic scars and keloid scars is thin and lacks normal skin markings and appendages. Common sites include the earlobe, upper chest, upper back, and deltoid region. They are rare on the mucous membranes, palms, soles, penis, scrotum, and eyelids.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


