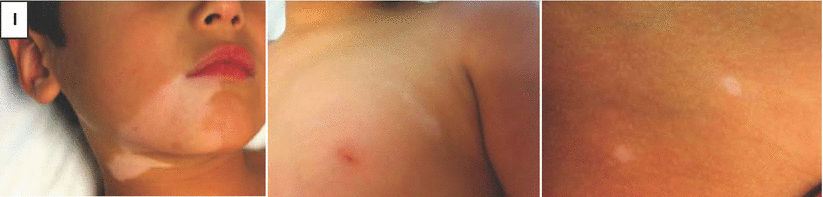
Fig. 28.1
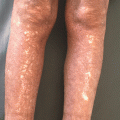
Clinical manifestations of vitiligo. (a) Patchy depigmentation on the hands. (b) Early macule of vitiligo on the arm. (c) Large, coalescing patches of vitiligo on the trunk. (d) Epidermal depigmentation on the knee, with sparing of the hair. (e) Poliosis from vitiligo on the scalp. (f) Erythema and scaling in a depigmented patch in a young girl with inflammatory vitiligo. (g) Unilateral depigmentation on the forehead of a young woman with segmental vitiligo. (h) Depigmentation on the shins of an avid mountain-biker, reflecting koebnerization of skin trauma. (i) Trichrome pigmentation in a patch of vitiligo that includes normal skin and depigmented skin with a hypopigmented border. (j) Perifollicular repigmentation from pigmented, but not depigmented, hairs on the abdomen of a patient receiving nbUVB phototherapy. (k) Near-complete repigmentation in a large patch on the abdomen of a patient receiving nbUVB phototherapy. (l) Mixed vitiligo with depigmentation in a well-defined unilateral segment on the right chin as well as remote areas of depigmentation on the left chest and right hip
A subset of patients have segmental vitiligo, which is characterized by depigmentation only on one side of the midline (Fig. 28.1g). During embryonic development, keratinocytes (ectoderm) and melanocytes (neural crest) emerge from the dorsal midline primitive streak, proliferate (keratinocytes) or migrate (melanocytes) ventrally, and generally meet at the midline, without crossing to the contralateral side [5]. This unilateral pattern of skin formation in the embryo is readily apparent in diseases of somatic mosaicism, where affected skin is unilateral and limited by the midline [5–8]. T cells, which are bone marrow-derived and recruited to the skin from the circulation, are thought to move freely through tissues, unlimited by the midline (reviewed in [9]). Therefore, the unilateral nature of segmental vitiligo should most likely be attributed to skin cells, rather than immune cells, and a form of somatic mosaicism could induce a unique susceptibility of melanocytes within a unilateral, focal segment to autoimmune attack. A recent study compared patterns of segmental vitiligo to those of known diseases of somatic mosaicism, and found that patterns of segmental vitiligo most resembled those of melanocytic origin [10, 11].
Disease Course
While it has a primarily progressive course, vitiligo can wax and wane, or even arrest altogether. Trauma to the skin may induce depigmentation at the site, a phenomenon known as koebnerization (Fig. 28.1h). Rapid spread of disease may occur, which has been associated with the trichrome variant in which three colors are evident, including the normal skin, depigmented skin, and an intervening zone of hypopigmentation that typically progresses to depigmentation (Fig. 28.1i). The inflammatory variant described above also typically marks rapidly progressive disease. Arrest of depigmentation, either spontaneous or induced, enables melanocytes to regenerate from pigmented hair follicles, a potential reservoir of melanocyte stem cells (Fig. 28.1j). Over time, these melanocytes can repigment the epidermis and erase any evidence of its existence [12] (Fig. 28.1k). In the segmental variant, depigmentation is usually limited to a focal, unilateral area, but may rarely spread beyond this initial site, a variant described as “mixed” vitiligo (Fig. 28.1l) [13].
Epidemiology and Diagnosis
Vitiligo affects approximately 0.5–1 % of the general population [14], although isolated populations where their genetics are more homogenous report higher rates [15]. The average age at diagnosis is 24 years [16], and half the patients who develop vitiligo do so before the age of 20. The segmental variant of vitiligo comprises ~30 % of childhood disease and 10–15 % of adult vitiligo, because childhood onset is more likely to be segmental [17]. The inflammatory variant of vitiligo is uncommon, and Vogt-Koyanagi-Harada Syndrome is very rare, although it appears to be most prevalent in Japan.
Vitiligo is typically a clinical diagnosis, and Woods lamp examination helps to distinguish the depigmentation of vitiligo from hypopigmentation of other conditions. The differential diagnosis of depigmentation is limited, and includes nevus depigmentosus, piebaldism, idiopathic guttate hypomelanosis (IGH), and trauma-induced depigmentation (burns and other scars). Nevus depigmentosus and piebaldism are present at or shortly after birth and grow with the child. IGH is characteristically limited to macules of small size on sun-exposed surfaces, and trauma-induced depigmentation is typically geographic-appearing and often recalled by the patient. If the diagnosis is not clinically clear, the lack of melanocytes can be confirmed through biopsy of the lesion using normal skin for comparison, although biopsy is rarely necessary. The differential diagnosis of hypopigmentation is extensive, and thus distinguishing depigmentation from hypopigmentation via Woods lamp examination is very helpful. Important hypopigmented conditions not to miss include hypopigmented mycosis fungoides due to the need for more intensive monitoring, lichen sclerosus et atrophicus (LSetA) due to the need for aggressive treatment to avoid permanent sequelae, as well as tinea versicolor, pityriasis alba, and progressive macular hypomelanosis due to simple alternative treatment strategies.
The risk of developing vitiligo in first-degree relatives of patients is 6.1 %, and in an identical twin is 23 % [16], implying a moderate genetic contribution to disease. The remainder of the risk has been described as environmental [18], which will be discussed below. A third possible explanation for twin disconcordance in vitiligo and other autoimmune diseases that is often not considered is a stochastic contribution, or risk conferred by chance during development. Stochastic influences occur during cell replication, differentiation, and migration during embryogenesis [19], the generation of T cell receptors through VDJ recombination and nucleotide insertion/deletion [20], and central tolerance induction in the thymus [21].
Psychological Impact
Vitiligo is a disfiguring condition, as skin color is an important component of identity. The porcelain-white lesions are brandished on the skin as if a public announcement of disease. Maybe for this reason, or for its similarity in appearance to tuberculoid leprosy (hypopigmented patches), vitiligo sufferers in India are frequently bypassed as candidates for arranged marriage [22]. Psychological consequences of vitiligo are severe, leading to depression, anxiety, sleep disturbances, sexual dysfunction, feelings of discrimination, and even suicidal thoughts and attempts. These emotional disturbances are comparable to those suffering from psoriasis and eczema [1, 23]. Public concerns over transmissibility of vitiligo make understanding its pathogenesis not only critical for the development of effective treatments to help our patients, but also for public education and acceptance of those who have been ridiculed and ostracized from their communities, beginning over 3500 years ago and continuing to this day [22].
Current Treatments
Treatments for vitiligo can be very effective for some patients, although they are all used “off-label”, as there are currently no treatments that are FDA-approved to promote repigmentation of vitiligo. Options include topical steroids, topical calcineurin inhibitors, and narrow-band ultraviolet light B (nbUVB) [24].
Studies reveal that nbUVB is the most effective single therapy for vitiligo, followed by ultrapotent topical steroids and calcineurin inhibitors [25]. Combined therapy with nbUVB and topicals has greater efficacy then either one alone. In a head-to-head comparison, nbUVB showed similar efficacy to psoralen with ultraviolet light A (PUVA), however nbUVB resulted in a better color match of repigmented skin to normal skin [26], and long-term treatment with nbUVB appears to be safe, while PUVA increases the risk of skin cancer [27]. Therefore, PUVA therapy is best avoided for vitiligo in favor of nbUVB. The frequency of treatment for nbUVB is most efficacious at 2–3 visits per week, as once weekly treatment has only modest efficacy. It appears that visits 3 times weekly induces repigmentation more rapidly than twice weekly, however over the long-term both schedules appear to have equal efficacy. Therefore after 2–3 months of treatment, those receiving therapy 3×/week typically have more repigmentation, while after six months of therapy either 2 or 3×/week have similar repigmentation. If possible for the patient, we often treat 3×/week for the first three months, and then decrease to 2×/week for the remainder of treatment.
Ultrapotent topical steroids appear to have slightly better efficacy than topical calcineurin inhibitors; however, they also have increased risk of epidermal atrophy, striae, and steroid-induced acne. Tacrolimus ointment does not induce atrophy, but may burn shortly after application and, in some patients, results in flushing of the skin after alcohol consumption. Burning seems to go away with repeated treatment. Skin flushing in public may be avoided by taking a small amount of alcohol to induce the flushing in a private environment, which then resolves and may not reappear with additional consumption. We often suggest patients treat with an ultrapotent topical steroid twice daily for one week, followed by tacrolimus ointment twice daily for one week. The patient then alternates treatment with steroid and calcineurin inhibitor each week, which appears to have good efficacy with significantly reduced risk of side effects.
In select cases where depigmentation is stable, autologous melanocytes can be harvested from uninvolved skin and transplanted to depigmented skin, resulting in repigmentation, a procedure called melanocyte-keratinocyte transplantation (MKTP) [17]. This approach may be curative for patients with highly stable disease, and is most useful for those with segmental vitiligo, a variant in which depigmentation is highly stable.
Avoidance of environmental triggers may prevent or reduce the severity of vitiligo. This includes avoiding exposure to phenol-containing products such as hair dyes, cleaning products, adhesives, and treated rubber products. Patient history can be helpful to identify exposures, though difficulty with recall and the large number of common household products involved can make this approach difficult. One study reported that patients with chemical-induced vitiligo often present with confetti-like macules of depigmentation, which may help to identify those patients in whom a detailed exposure history should be taken [28].
Monobenzyl ether of hydroquinone, or monobenzone, was first implicated as a risk factor for vitiligo through occupational exposure in a group of factory workers in 1939 [29]. It is now the only FDA-approved treatment for vitiligo, used for removing the remaining pigment in a patient with widespread disease. Its mechanism is through the exacerbation of vitiligo, described below. For clinical use, the chemical is compounded as a 20 % ointment and should be applied daily to pigmented skin. Successful depigmentation can take up to 12 months, and should be considered irreversible. In addition, pigmented skin remote from the application site may also depigment, so this strategy should not be used for localized depigmentation. A subset of those who use this approach exhibit allergic contact dermatitis to the chemical, which may be treatment-limiting. The majority of those who successfully undergo depigmentation with monobenzone topical therapy are very pleased with the results [30].
Pathophysiology of Vitiligo in Humans
Historically it has been debated whether vitiligo is an autoimmune disease or a melanocyte-intrinsic degenerative syndrome [31]. Like most other autoimmune diseases, vitiligo is multifactorial, requiring both genetic and environmental factors for melanocyte destruction. Melanocytes from vitiligo patients are difficult to grow in vitro, are more susceptible to oxidative stress, and have elevated cellular stress responses [32]. Autoimmunity plays a key role in pathogenesis as well, evident by the fact that the majority of genes that confer susceptibility to vitiligo play central roles in immune responses, and “adoptive transfer” of vitiligo has been observed in patients receiving bone marrow/stem cell transplants from donors with vitiligo [33–35]. In this section we will outline the pathophysiology of vitiligo with an emphasis on the recent literature.
Melanocyte Stress
Melanocytes are particularly susceptible to cellular stress, due to their exposure to UV irradiation and other environmental stressors, as well as the production of melanin, which generates reactive oxygen species (ROS) and activates the unfolded protein response (UPR). Electron microscopy of cellular substructure revealed that the endoplasmic reticulum (ER) in melanocytes from vitiligo patients was dilated compared to healthy controls [36], a characteristic that implicates cellular stress. Elevated levels of H2O2 and oxidative byproducts are present in vitiligo patients’ epidermis compared with controls, suggesting uncontrolled generation of reactive oxygen species (ROS) [37]. In addition the enzyme catalase, which reduces H2O2 to O2 and H2O and thereby relieves oxidative stress, is decreased in lesional skin [38], which may be either a cause or an effect of increased H2O2. Treatment of vitiligo with topical exogenous catalase has been attempted, with variable results [39].
Tyrosinase facilitates a number of steps that convert the amino acid tyrosine to melanin. Tyrosine is a phenol, containing a hydroxyl group attached to a benzene ring. Monobenzone was the first chemical reported to induce vitiligo in a group of factory workers exposed to the chemical through acid-cured rubber gloves. Distant spread of depigmentation to areas beyond the exposure sites (hands and forearms) suggested that monobenzone is not simply directly cytotoxic to melanocytes, but induces autoimmunity during exposure. Monobenzone treatment of vitiligo patients, used to depigment their skin to induce an even tone, also induces depigmentation at sites remote from the site of application, supporting this concept. Like tyrosine, monobenzone and other depigmenting agents are phenols, which act as tyrosine analogs but bind irreversibly to tyrosinase and thereby interrupt the melanin synthesis pathway [40]. This induces cellular stress through the induction of ROS and activation of the UPR, which then results in activation of innate immunity to induce inflammation [40–42]. Many household products contain other chemical phenols, including hair dyes and cleaning products, which have both been reported to induce chemical leukoderma, or chemically-induced vitiligo [28]. It is likely that phenols used as ingredients in these products act in a similar way as monobenzone, producing cellular stress through interaction with tyrosinase.
Initiation/Innate Immune Activation
There is evidence that the initiation of vitiligo requires innate immune activation. Several studies have implicated innate immune sensing of cellular stress, including dendritic cell (DC) activation by stress-induced release of exosomes [40], the heat shock protein HSP70i [43] and an apparent increased recruitment of natural killer (NK) cells into the skin of vitiligo patients [44]. Melanocyte stress is therefore linked to innate immune activation, and thus initiation of inflammation that may lead to adaptive autoimmunity; however, more studies will be required to identify the precise signaling pathways involved [45]. Innate immune gene polymorphisms in vitiligo have been identified through genome-wide association studies, though their functional roles in disease are still being defined (see section “Genetic Contributions”). Environmental triggers that induce melanocyte stress have been identified, and patients may be counseled to avoid these exposures (see section “Melanocyte Stress”).
Adaptive/Autoimmune Response
Vitiligo is driven by CD8+ T cell-mediated killing of melanocytes. This has been demonstrated ex vivo using a human skin culture system in which melanocyte-specific T cells isolated from affected skin migrated into an unaffected skin explant from the patient and induced melanocyte apoptosis. CD8+ T cells were both necessary and sufficient for this effect [46]. Increased frequencies of melanocyte-specific CD8+ T cells in the blood and skin correlate with disease severity [47]. The antigen specificities of the T cell receptors (TCRs) in vitiligo have been identified and are shared with melanoma, including MART-1, gp100, tyrosinase, and tyrosinase-related proteins 1 & 2 (TRP1 & 2) [48]. These T cells produce IFNγ which, in addition to inducing T cell recruitment, may be directly cytotoxic to melanocytes [49]. It is possible that vitiligo results from an overzealous anti-tumor response [50]. Patients with vitiligo are at a three-fold lower risk for melanoma, and those with melanoma who spontaneously regress or who are cured following treatment frequently develop vitiligo [51].
CD4+ helper T cell responses may also play a role in vitiligo via IFNγ production, although functional evidence for their role is lacking [52, 53]. There is growing evidence that vitiligo is dependent on IFNγ and downstream genes including IFNγ-inducible chemokines [52, 54]. There are several case studies of patients who were treated with IFNα for hepatitis who developed vitiligo [55–64], suggesting that type I interferons can induce disease, though it is unclear what their role is in spontaneous vitiligo. Several studies have questioned whether TH17 cells and IL-17 drive pathogenesis, but their functional role in vitiligo remains unclear. It is possible that a subset of vitiligo patients, such as those with inflammatory vitiligo, also have an IL-17 component in their disease pathogenesis, but studies to date have not classified TH1 versus TH17 in different vitiligo subtypes. Autoantibodies against melanocytes have been identified in patients [65], though it is uncertain whether they play a direct role in pathogenesis or are merely a biomarker of disease. A role for regulatory T cells (Tregs) have also been implicated in vitiligo through genetic studies [66] and the fact that patients with immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome, who lack Tregs, have an increased incidence of vitiligo [67]. However evidence is conflicted regarding whether their numbers are normal or fewer, whether they have difficulty homing to the skin, and whether or not they are functionally impaired [68–72].
Hair depigmentation is not commonly observed in vitiligo, most likely due to hair follicle immune privilege. Immune privilege is defined as tissue that is not readily inflamed, possibly due to limited access by the immune system and/or the expression of immunoregulatory proteins or cell populations [73–75]. Melanocyte stem cells reside in hair follicles, which migrate out into the epidermis to repigment the skin [76]. When repigmentation occurs in patients, it usually begins around pigmented hair follicles and spreads outward, indicating that the stem cell populations in the hair follicles may be maintained through immune privilege. Little is known about the mechanisms of repigmentation, though this is critical for effective therapy (see also section “Future Perspectives into Vitiligo Treatment Strategies”). A summary of vitiligo pathogenesis is presented in Fig. 28.2.
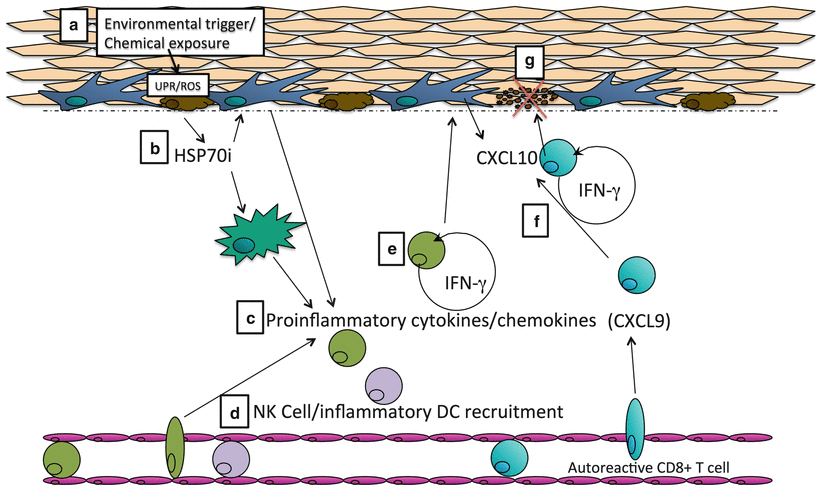
Fig. 28.2
Vitiligo pathogenesis: linking melanocyte stress, innate immune triggering and adaptive immune responses. (A) Exposure to an environmental insult, such as a phenol, may induce stress responses in melanocytes through activation of the unfolded protein response (UPR) or production of reactive oxygen species (ROS). (B) This results in release of stress/danger signals such as HSP70i, which are detected by other skin resident immune cells such as Langerhans cells or dermal dendritic cells. (C) These cells secrete proinflammatory cytokines and chemokines to recruit more immune cells to the skin, including natural killer (NK) cells or inflammatory dendritic cells (DCs). (D) NK cells produce IFN-γ, which stimulates CXCL10 production by skin resident cells (E). (F) Established gradients of CXCL9 and CXCL10 from the dermis to the epidermis direct autoreactive CD8+ T cell recruitment to the skin and up to the epidermis, where they kill melanocytes either through cell contact dependent mechanisms or through IFN-γ production, which induces additional chemokine production to promote the cycle (G)
Genetic Contributions
Genetic contributions to vitiligo are well-established, based on familial associations of the disease, as well as the increased concordance in identical twins [16, 66]. It is likely that inherited factors affect all pathways involved in vitiligo pathogenesis, including melanocyte stress, innate inflammation, and adaptive autoimmunity. The earliest genetic associations with vitiligo were with the HLA-A haplotypes, required for T cell recognition of their target cells [77]. The first non-HLA gene found to confer risk for vitiligo was NACHT, LRR and PYD domains-containing protein 1 (NLRP1). NLRP1 is a component of the innate immune response, confirming a causative role of innate immunity in vitiligo [78]. A recent study using functional genomics demonstrated that NLRP1 haplotypes associated with vitiligo cause increased IL-1β secretion by monocytes, potentially predisposing patients to innate immune activation and lowering the tolerance threshold [79].
Genome-wide association studies (GWAS) have revealed a large number of additional genes, the majority of which also play roles in immune responses (PTPN22, TSLP, HLA class I/II/III, CCR6, IL2RA, UBASH3A, and FOXP3), including specifically cytotoxic T cell responses (GZMB), supporting earlier histologic and in vitro mechanistic data [18]. An allele of tyrosinase, the enzyme responsible for creating melanin, is protective against melanoma but causes an increased risk of vitiligo [80, 81]. X-box binding protein 1 (XBP1) variants were also identified as risk factors for vitiligo [82]. XBP1 promotes class II HLA expression (important for antigen presentation to CD4 T cells) [83], IL-6 and IL-8 production [84], and it is also a member of the UPR that protects the cell from stress-induced apoptosis [85].
In addition to a proinflammatory role in vitiligo, XBP1 may also participate in the stress response, as decreased expression or impaired activity of the protein may increase stress levels of the melanocyte [85], indirectly leading to activation of autoimmunity. Consistent with this hypothesis, studies in inflammatory bowel disease have identified two hypomorphic variants of XBP1 that contribute to a proinflammatory state of the intestine as a result of increased cellular stress [86]. To date, only a hypermorphic variant has been described in vitiligo [87], which may induce an exaggerated inflammatory response to cellular stress [41]. Another stress-related genetic factor that has been identified is tyrosinase-related protein 1 (TRP1). Mutations in TRP1 have been identified in vitiligo patients, and have been shown to activate cellular stress responses in melanocytes via chaperone proteins such as calnexin [32]. Further studies will be required to determine the functional role of other identified genetic factors in disease pathogenesis (Fig. 28.3).
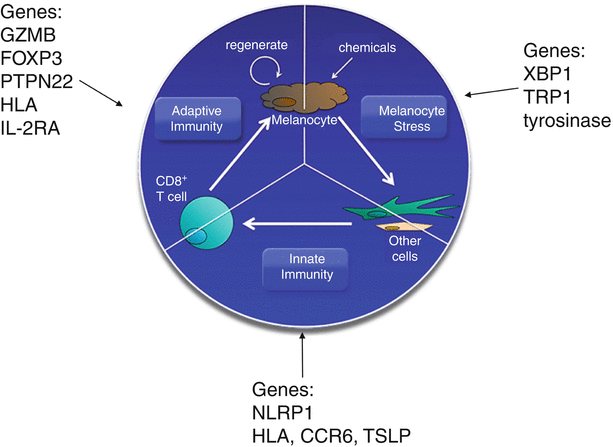
Fig. 28.3
Multiple pathways contribute to vitiligo pathogenesis. Melanocyte stress can be induced by chemicals and other environmental insults, resulting in release of stress signals that are detected by other cells in the skin. This can trigger activation of the innate immune system, resulting in production of chemokines required for autoreactive CD8+ T cell recruitment to the skin. These CD8+ T cells target and kill melanocytes, resulting in release of more proinflammatory signals in the skin, thereby re-activating the cycle. As melanocytes try to regenerate, they may continue to be targeted by CD8+ T cells in the skin, resulting in maintenance of clinical depigmentation. Examples of genes that may directly affect these three major pathways in disease are highlighted
Basic and Translational Approaches for Studying Vitiligo
Basic Science Approaches: Animal Models of Disease
One of the earliest models of vitiligo identified was a spontaneous mouse model caused by a point mutation in the microphthalmia-associated transcription factor (MITF) gene, which caused melanocyte degeneration as the mouse aged. However due to its monogenic nature and lack of autoimmunity [88, 89], this strain (mivit) does not appear to accurately model human vitiligo pathogenesis. Several other mouse models have been developed to study vitiligo, including models induced through exposure of the skin to melanocyte antigens plus adjuvant to induce depigmentation [90], or through adoptive transfer of transgenic T cells engineered to react against melanocyte antigens. Early models were developed from animal models of melanoma immunotherapy [91–95] in which TCR transgenic mice responding to gp100 (called PMEL for pre-melanosome protein were used to mediate tumor clearance. The mice spontaneously developed white hair, and several studies have used these PMEL host mice to study spontaneous hair depigmentation [43]. We developed a model through the adoptive transfer of PMEL T cells into hosts that have epidermal melanocytes [95], which resulted in progressive epidermal depigmentation with sparing of the hair (Fig. 28.4a). Studies using this model implicated IFN-γ and the IFN-γ-induced chemokine CXCL10 as required for both disease progression and maintenance [54]. This model is most useful for studying the effector phase of vitiligo, as loss of tolerance through innate immune activation is largely bypassed.
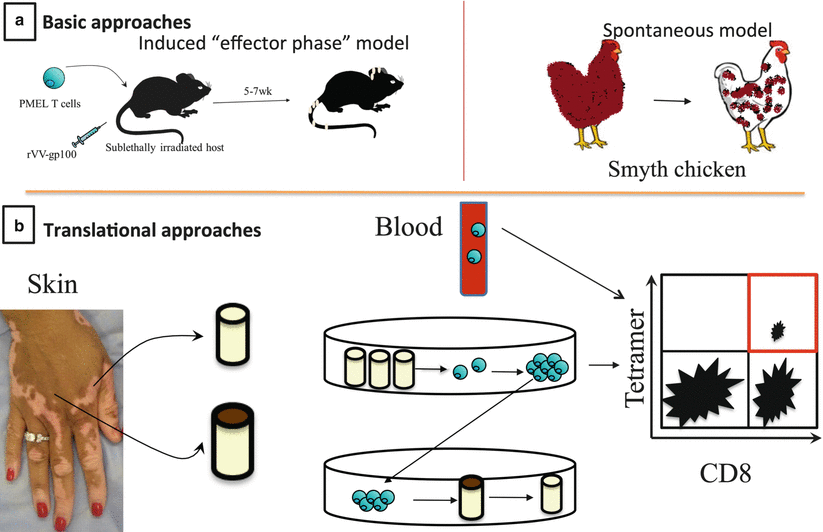
Fig. 28.4
Experimental approaches for studying vitiligo. (a) Basic science approaches for studying vitiligo: animal models. Adoptive T cell transfer model for studying the effector phase of vitiligo in mice. Melanocyte antigen-specific T cells (PMEL) are adoptively transferred into hosts and activated in vivo with an attenuated recombinant vaccinia virus expressing their cognate antigen gp100 (rVV-gp100). After 5–7 weeks, mice develop patchy depigmentation. This model is most useful for studying the “effector phase” of vitiligo. Alternatively, the Smyth Line Chicken spontaneously develops vitiligo, and is a more appropriate model for studying induction of vitiligo. (b) Translational approaches for studying vitiligo. Human samples can be analyzed ex vivo to examine elements involved in disease pathogenesis. Using skin biopsies, antigen-specific T cells can be isolated from lesions, cultured, expanded, and used to examine killing ability in normally pigmented skin cultures [46]. Antigen specificities of these skin T cells as well as T cells isolated from peripheral blood of vitiligo patients can be analyzed with tetramers, which are fluorescently labeled reagents comprised of MHC:peptide complexes. Most of the melanocyte tetramer reagents available are for CD8+ T cells, though it is possible that reagents for CD4+ T cells will become available in the future
Perhaps the most representative animal model of vitiligo is the Smyth Line (SL) chicken, which develops spontaneous depigmentation of its feathers over time (Fig. 28.4a). Affected brown SL chickens develop white feathers with age and produce anti-melanocyte antibodies. Depigmentation is due to melanocyte loss by apoptosis induced by cytotoxic T cells. Interestingly, the SL chicken also has melanocyte defects, including autophagocytosis of melanosomes and increased generation of ROS. Consistent with the theory that vitiligo pathogenesis is multifaceted, these melanocyte defects are not sufficient for depigmentation in the absence of a functional immune system [96]. While the SL chicken appears to model vitiligo in a way that is most similar to human disease, the paucity of tools and expertise for investigation of chicken cells limits its use. Recent studies determined that IFNγ, IL-21 and IL-10 are expressed in evolving lesions [97], and microarray analysis has been conducted to begin to address gene signatures in the model [98].
Translational Approaches Using Patient Samples
Histology and immunohistochemistry were the earliest methods used to study inflammatory infiltrates in vitiligo. Histologic analysis of vitiligo patient biopsies revealed a lack of epidermal melanocytes with accompanying lymphomonocytic infiltrates. Immunohistochemistry revealed infiltrates of CD8 and CD4 T cells that are most often detected perilesionally [3, 99, 100]. CD8 T cells have been observed infiltrating the epidermis and may be found proximal to dying melanocytes. Innate immune populations including CD11b+ CD11c+ inflammatory DCs [43] and NK cells [44] have also been identified in vitiligo lesions.
Van den Boorn et al. used human skin organ culture and autologous T cells to demonstrate that CD8+ T cells are necessary and sufficient to induce melanocyte apoptosis in human skin, consistent with a causative role for adaptive immunity in vitiligo [46]. This approach models processes that occur within the skin, and provides the normal 3D architecture and maintains most of the microenvironment. Punch biopsies of lesional skin from vitiligo patients were cultured ex vivo, T cells migrated out of the explant, and were expanded in vitro. Expanded populations can then be analyzed, manipulated, and re-exposed to punch biopsies of normally pigmented skin from the same patient to determine their effects on melanocytes in situ (Fig. 28.4b). Disadvantages of this approach include the inability to model recruitment of T cells from the blood to the tissue, and the fact that over time culture of skin results in the loss of resident immune cell populations through emigration [101].
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


