Autoinflammatory disease
Inheritance
Gene/Protein
Age of onset
Flare duration
Mucocutaneous manifestations
Musculoskeletal manifestations
Systemic manifestations
Treatment
FMF
AR
MEFV/pyrin
First three decades of life
1–3 days
Erysipeloid-like rash on dorsal hands and feet
Arthritis
Fever
Polyserositis
AA amyloidoisis
Colchicine
IL-1 antagonists
TRAPS
AR
TNFRSF1A/TNFR1 receptor
Childhood or adolescence
7–14 days
Migratory erythema
Periorbital edema
Conjunctivitis
Arthralgia, arthritis
Fever
Pleuritis
Abdominal pain
AA amyloidosis
TNFα antagonists
IL-1 antagonists
MKD
AR
MVK/mevalonate kinase
First year of life
3–7 days
Urticaria-like eruption
Morbiliform eruption
Petechiae
Vasculitic purpura
Arthralgia, arthritis
Fever
Cervical lymphadenopathy
Abdominal pain
Vomiting, diarrhea
Splenomegaly
NSAIDs
TNFα antagonists
IL-1 antagonists
FCAS
AD
NLRP3/CIAS1/NLRP3
Usually in childhood
24 h
Cold-induced neutrophilic urticaria
Conjunctivitis
Arthralgia
Fever
Rare secondary amyloidosis
IL-1 antagonists
MWS
AD
NLRP3/CIAS1/NLRP3
Childhood
1–3 days
Neutrophilic urticaria
Conjunctivitis
Uveitis
Scleritis
Arthralgia
Fever
Sensorineural deafness
Papilledema
Aseptic meningitis
AA amyloidosis
IL-1 antagonists
NOMID
AD
NLRP3/CIAS1/NLRP3
First weeks of life
Continuous
Neutrophilic urticaria
Conjunctivitis
Uveitis
Erosive arthritis
Joint deformities
Fever
AA amyloidosis
Papilledema
Aseptic meningitis
Seizures
Cognitive impairment
Sensorineural deafness
IL-1 antagonists
DIRA
AR
IL1RN/IL1RA
At birth or within first weeks of life
Continuous
Sterile pustulosis
Oral ulcerations
Joint swelling
Hyperostosis
Osteolysis
Low-grade fever
Premature birth
HSM
Respiratory insufficiency
Thrombosis
Anakinra
DITRA
AR
IL36RN/IL36RA
Childhood, adulthood,
Peri-partum
Days to weeks
Sterile pustulosis
Arthralgias
Fever
Acitretin
IL-1 antagonists
SAPHO/CRMO
Unknown
Unknown
Childhood or young adulthood usually
Weeks
Palmoplantar pustulosis (60 %)
Acne (25 %)
Neutrophilic dermatoses
Hyperostosis
Osteolysis
Synovitis
Fever
NSAIDs
Bisphosphonates
Methotrexate
TNFα antagonist
IL-1 antagonists
Majeed syndrome
AR
LPIN2/lipin-2
Neonatal
Continuous
Neutrophilic dermatoses
Hyperostosis
Osteolysis
Congenital dyserythropoetic anemia
IL-1 antagonists
CAMPS
AD
CARD14/CARD14
Infancy
Continuous
Generalized pustulosis
Arthralgia
Fever
IL-12/23 antagonist
PAPA
AD
PSTPIP1/PSTPIP1
Childhood
Weeks
Pyoderma gangrenosum
Acne
Pyogenic arthritis
Fever
TNFα antagonist
IL-1 antagonists
PRAAS
AR
PSMB8/PSMB8
First weeks of life
Days to weeks
Violaceous annular plaques
Heliotrope rash, periorbital swelling
Lipodystrophy
Variable arthritis
Myositis
Fever
HSM
Microcytic anemia
Lymphadenopathy
Metabolic abnormalities
No established therapy
PLAID
AD
PLCG2/phospholipase Cγ2
Early childhood
Hours
Cold-induced urticaria
Atopy
Granulomatous rash
None
Autoimmune thyroiditis Sinopulmonary infection
No established therapy
APLAID
AD
PLCG2/phospholipase Cγ2
Infancy
Continuous, triggered by sun and heat exposure
Recurrent vesiculobullae
Corneal bullae and ulcerations
Arthralgia
Recurrent sinopulmonary infection
Enterocolitis
No established therapy
Corticosteroids, IL-1 antagonists have been used
DADA2
AR
CECR1/ADA2
Early childhood
Continuous
Livedo racemosa
None
Fever
Vasculopathic changes
HSM
Ophthalmologic involvement
Mild immunodeficiency
No established therapy
Innate Immunity
The innate immune system provides an immediate and nonspecific host defense against infection while the adaptive immune system provides a more complex, antigen-specific immune response. Effector cells of innate immunity include phagocytes, such as macrophages, dendritic cells and other antigen presenting cells, whereas in autoimmune diseases, B and T lymphocytes are the primary mediators of the inflammatory response.
The innate immune system acts through pattern recognition receptors (PRR) which recognize highly conserved pathogen motifs called pathogen-associated molecular patterns (PAMPs) and damage motifs known as damage-associated molecular patterns (DAMPs). There are three recognized types of PRRs employed by the innate immune system: Toll-like receptors (TLRs), NOD-like receptors (NLRs) and retinoic-acid-inducible-gene-1-like receptors (RLRs). Recognition of foreign material by PRRs leads to activation of signal transduction pathways which signal gene expression of pro-inflammatory cytokines, including interleukin-1 (IL-1) family cytokines, interferon α (IFNα), interferon γ (IFNγ), and tumor necrosis factor α (TNFα). Chronic, prolonged, unmitigated, excessive activation of PRRs can lead to autoinflammation and autoinflammatory diseases [2].
The Inflammasome
The first-described autoinflammatory diseases, the classic periodic fevers syndromes and the cryopyrin-associated periodic syndromes (CAPS) – were characterized by dysregulation of inflammasome activation. Activation of the NLRs leads to the formation of large multimeric complex protein structures, known as inflammasomes, which are critical for host defense against infection. Two main inflammasomes have been described: the NALP1 inflammasome and the NALP3 inflammasome, also known as the cryopyrin inflammasome [3]. In response to microbial components or endogenous metabolic stress, inflammasomes mediate procaspase activation which in turn catalyzes the cleavage of pro-IL-1β and pro-IL-18 into activated IL-1β and IL-18, respectively. IL-1β is produced primarily by myeloid cells and is the primary cytokine implicated in several autoinflammatory diseases. The IL-1 receptor (IL-1R) is ubiquitously expressed, and binding of IL-1β to the IL-1R results in proinflammatory signaling through NFkB-mediated transcription of proinflammatory genes [4]. The most well-described inflammasome, the NALP3 inflammasome, plays a key role in the pathogenesis of Familial Mediterranean Fever (FMF), the TNF receptor-associated periodic syndrome (TRAPS) and CAPS [3] (Fig. 40.1).
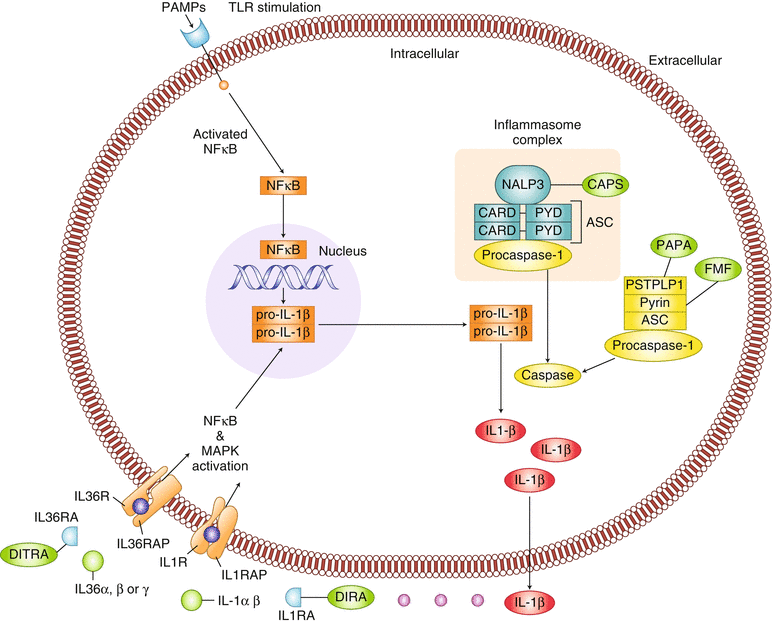
Fig. 40.1
Pathogenic mechanisms of autoinflammatory diseases. PAMP pattern recognition receptor, TLR toll-like receptor, NALP3, NACHT LRR and PYD domains-containing protein 3 (NALP3) or cryopyrin, CARD caspase recruitment domain, PYD pyrin domain, ASC apoptosis-associated speck-like protein containing a CARD, PSTPIP1 proline serine threonine phosphatase interacting protein 1, IL-1R IL-1 receptor. IL-1RAP IL-1 receptor accessory protein, IL-36R Il-36 receptor, IL-36RAP IL-36 receptor accessory protein, CAPS cryopyrin associated periodic fever syndrome, PAPA pyogenic arthritis, pyoderma gangrenosum, acne. FMF familial Mediterranean fever, DIRA deficiency of the IL-1 receptor antagonist, DITRA deficiency of the IL-36 receptor antagonist
Since the initial characterization of the cryopyrin-associated fever syndromes, numerous other monogenic autoinflammatory disorders have been described, including those which lead to pustular and other neutrophilic skin manifestations, and which further contribute to our understanding of IL -1 and non-IL-1-mediated innate immune pathways driving inflammatory skin disease. Characterization of several monogenic diseases has also led to use of targeted therapeutics for their management. Importantly, these insights have also helped to begin to dissect pathogenic mechanisms of phenotypically similar autoinflammatory diseases with as yet no known genetic etiology, and identify appropriate targeted therapeutics for their management, thereby improving human health.
This chapter will introduce monogenic autoinflammatory diseases and discuss pathogenic mechanisms, clinical features and therapeutic options for these disorders.
Classic Fever Syndromes
Familial Mediterranean Fever
The term Familial Mediterranean Fever (FMF) was first proposed in 1958 to describe a periodic fever syndrome which predominated in individuals of Eastern Mediterranean descent [5]. It is the most prevalent autoinflammatory disease worldwide [6]. Although FMF is classically recessively-inherited [7, 8], dominant inheritance and clinically symptomatic heterozygotes have been reported [9]. Genetic mutations in the MEFV gene are responsible for FMF and were first reported in 1997 [7, 8]. Almost one-half of the mutations described to date have been identified on exon 10, underscoring the importance of this exon in the function of MEFV [10].
The MEFV gene encodes the protein pyrin which binds the apoptotic speck (ASC) adaptor protein in the NALP3 inflammasome. Binding of wildtype pyrin and ASC inhibits assembly of the NALP3 inflammasome [11]. Mutations in MEFV are gain-of-function mutations affecting pyrin, thereby leading to increased inflammasome activity and subsequent IL-1β production. This hypothesis has been confirmed in a pyrin knock-out murine model as well as a murine model with truncated pyrin protein in which increased caspase-1 activation and increased IL-1β maturation is observed [12]. In a pyrin knock-in murine model, it was demonstrated that pyrin could form an inflammasome of its own, independent of NALP3, which could activate proinflammatory cytokine IL-1 [13].
Individuals with FMF typically present within the first three decades of life with acute episodes of fevers, erysipelas-like rash, arthritis and polyserositis. Flares can last 1–3 days in duration, and occur as frequently as weekly or as rarely as every few years [14, 15]. The erysipelas-like rash of FMF tends to involve the hands and dorsal feet, lasting up to 72 hours and recovering with recrudescence of fever (Fig. 40.2a). Approximately 30 % of affected individuals will experience acute nonerosive arthritis during flares. The majority of patients develop monoarthritis (70 %), and fewer develop oligo- (26 %) and polyarthritis (4 %). The most commonly affected joints include the knees, followed by ankles and hips. Arthritis is typically self-limited and not associated with permanent periarticular or cartilage damage [16]. Polyserositis can manifest as an acute abdomen, pleuritis, pericarditis, scrotal pain and more rarely, aseptic meningitis [17]. Gastrointestinal disease manifestations, ranging from mild abdominal pain to noninfectious peritonitis, occur in 95 % of FMF patients [18]. Prolonged myalgias in the setting of fevers [19], elevated inflammatory markers likely secondary to vasculitis [20], as well as HLA-B27-independent sacroiliitis have also been reported [21].
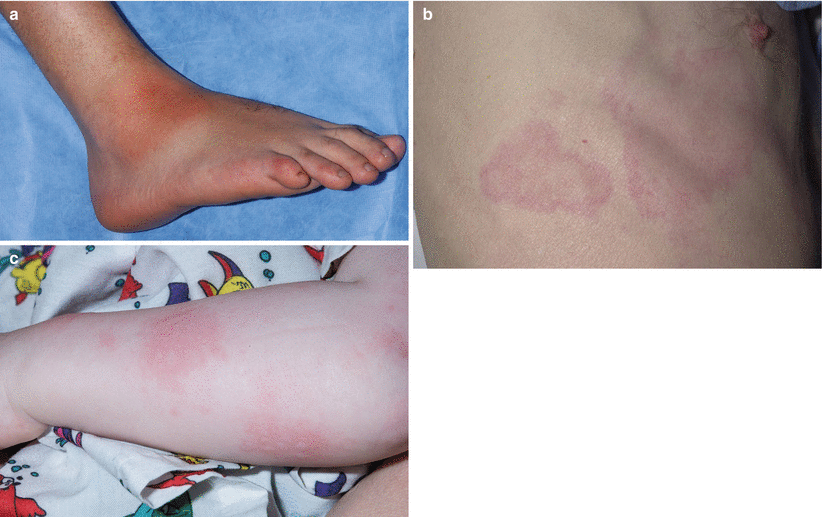
Fig. 40.2
Dermatologic manifestations of classic periodic fever syndromes. (a) Erysipeloid erythema involving the anterolateral ankle of a child with Familial Mediterranean Fever (FMF). (b) Migratory erythema with serpiginous borders and central clearing on the right lateral chest of a young man with TNF receptor-associated periodic syndrome (TRAPS). (c) Urticaria-like plaques involving the left arm of a child with mevalonate kinase deficiency (MKD)
Secondary AA amyloidosis is a prominent feature of chronic and uncontrolled FMF [22]. In the setting of chronic inflammation, AA amyloid is subject to protein misfolding and subsequent deposition in extracellular matrices of various tissues leading to organ impairment. In FMF, amyloidosis typically involves the kidneys. Affected individuals present with proteinuria and subsequently develop nephrotic syndrome and uremia, ultimately leading to renal impairment and death. Biomarkers for FMF include elevated serum amyloid A as well as elevated levels of DAMP protein S100A12. The latter has been shown to correlate with joint disease severity [23, 24]. Elevated systemic inflammatory markers have been during attacks as well as persist during attack-free periods [25].
Skin histopathology of FMF erysipelas-like erythema demonstrates superficial dermal edema and sparse perivascular infiltrate composed of neutrophils and few lymphocytes. Deposits of C3 in the vessel walls of the superficial vascular plexus can be seen on direct immunofluorescence [26–30]. Synovial fluid from affected joints ranges from non-inflammatory to septic-appearing [16].
Oral colchicine is the mainstay of therapy for the management of recurrent attacks and secondary amyloidosis associated with FMF [31]. In addition to achieving remission of inflammatory flares in the majority of patients, oral colchicine 1–2 mg daily has been associated with improvement of renal function in 95 % of affected individuals with proteinuria but not nephrotic syndrome. Furthermore, colchicine use has been shown to prevent the development of amyloid renal disease in FMF patients [16]. IL-1 antagonists should be considered in individuals who do not respond to or continue to have progressive renal amyloidosis despite oral colchicine therapy, as well as in patients who are intolerant to oral colchicine [32–36].
TNF Receptor-Associated Periodic Syndrome
Shortly after the discovery of the genetic cause of FMF, the genetic basis of TNF receptor-associated periodic syndrome (TRAPS) was reported in 1999 [37]. TRAPS is a dominantly-inherited disorder caused by mutations in the TNF receptor superfamily member 1A (TNFRSF1A) gene which encodes the TNFR1 receptor protein. Over 100 different mutations in TNFRSF1A have been reported to date [38].
The TNFR1 receptor is ubiquitously expressed. Mutations in TNFRSF1A lead to TNFR1 receptor protein misfolding. As a result, TNFR1 is unable to be transported to the cell membrane and therefore is sequestered in the endoplasmic reticulum at levels tenfold higher than the wild type protein [39, 40]. Mutations in TNFR1 lead to constitutive activation of MAP-kinases with subsequent uncontrolled production of IL-1 and TNFα.
The majority of TRAPS patients present in childhood or adolescence with a constellation of symptoms that can include recurrent fevers, abdominal pain, pleuritis, arthralgias, myalgias and periorbital edema and/or conjunctivitis. Attacks last for approximately 7–14 days but they may persist for up to 4 weeks. Abdominal pain occurs in 88 % of TRAPS patients, ranging from mild to moderate pain to acute abdomen [41]. Cutaneous manifestations occur in 69-87 % of TRAPS patients, most commonly as centrifugal migratory erythematous patches overlying sites of myalgia. Serpiginous patches and plaques and urticaria-like eruptions are less common [42] (Fig. 40.2b). The rash tends to progress from proximal to distal sites concurrent with myalgia symptoms. Arthralgia occurs in two-thirds of patients, involving peripheral joints in a monoarticular or oligoarticular pattern. Arthritis is less common [43, 44]. Periorbital edema is a hallmark feature of TRAPS, and conjunctivitis and uveitis has also been reported in approximately one-half of affected individuals [45]. Like FMF, renal deposition of AA amyloid, and subsequent renal impairment, is a prominent feature of TRAPS, affecting 8–10 % of patients with TRAPS [23]. Serum amyloid A (SAA) can be used as a biomarker in TRAPS [23].
Skin histopathology of the migratory rash associated with TRAPS is notable for superficial and deep perivascular and interstitial infiltrate of lymphocytes and monocytes. Small vessel vasculitis and recurrent panniculitis have also been reported [42]. Myalgias have been associated with monocytic fasciitis [46].
Definitive management of TRAPS remains elusive. Initially, it was thought that utilization of TNFα antagonists would be highly successful in ameliorating the signs and symptoms of disease. A prospective study demonstrated that etanercept, the soluable p75 TNFR:Fe fusion protein, reduced the frequency and intensity of attacks but did not lead to complete resolution [47]. Furthermore, a diminished effect of etanercept over time has been reported [47]. Paradoxically, anti-TNFα monoclonal antibodies have also been reported to cause an acute worsening of a patient’s clinical disease [48–51]. The IL-1 antagonists anakinra and canakinumab have also been used for the management of TRAPS with variable response [52, 53].
Mevalonate Kinase Deficiency
Mevalonate kinase deficiency (MKD) (formerly known as Hyperimmunoglobulinemia D and periodic fever Syndrome (HIDS) and Hibernian fever) is a recessively inherited condition caused by mutations in the mevalonate kinase (MVK) gene which encodes the mevalonate kinase protein [54, 55]. The most commonly reported mutation is V377I but however more than 30 distinct mutations have been reported [56].
Mevalonate kinase catalyzes the conversion of mevalonic acid to 5-phosphomevalonic acid in the biosynthesis of cholesterol and nonsterolisoprenoids. Mutations in MVK lead to decreased mevalonate kinase enzymatic activity which leads to reduction in isoprenoids, products of cholesterol biosynthesis pathways [57]. In vitro data from MKD patients demonstrate that isoprenoid biosynthesis leads to decreased IL-1β secretion in MKD leukocytes [58]; however, the mechanism by which depletion of isoprenoids results in increased circulating proinflammatory IL-1β is an area of ongoing investigation.
Affected individuals present within the first year of life with recurrent fevers, rash and arthralgias that can last 3–7 days and recur every 4–6 weeks. Flares can be triggered by childhood vaccinations, trauma, infection and stress. Cutaneous manifestations include an urticaria-like eruption, morbiliform eruption, petechiae and vasculitic purpura [59, 60] (Fig. 40.2c). Approximately two-thirds of affected individuals develop polyarthralgias and/or a nonerosive polyarthritis but it is not uncommon for only a single joint to be involved. The most commonly affected sites of joint involvement are large joints such as the knees and ankles [43, 44]. Bilateral cervical lymphadenopathy is a prominent feature of HIDS. Abdominal pain, vomiting, diarrhea, mucosal ulcerations and splenomegaly can also been seen. Unlike FMF and TRAPS, amyloidosis is an atypical finding [60].
Elevated serum ESR, CRP, IgA, IgD and urine mevalonic acid levels can be detected during disease flares. Urine mevalonic acid levels are currently used for diagnosis of MKD. Elevated IgD levels can also be seen in FMF and TRAPS and, therefore, should not be used for diagnosis [18]. Patients with MKD may also have normal IgD levels. Histologic examination of skin lesions reveal perivascular lymphocytic infiltrate, and may demonstrate vasculitic features and a neutrophilic infiltrate [59, 60].
IL-1 Family-Mediated Autoinflammatory Diseases
Cryopyrin-Associated Periodic Syndromes
Cryopyrin-associated periodic syndromes (CAPS), or the cryopyrinopathies, comprise a spectrum of 3 disorders characterized by autosomal dominant gain-of-function mutations in the NLRP3/CIAS1 gene which encodes the NLRP3 protein [64]. Ranging from mildest to most severe in presentation, the three disorders include familial cold-induced autoinflammatory syndrome (FCAS), Muckle-Wells syndrome (MWS) and neonatal onset multisystem inflammatory disorder/chronic infantile neurologic, cutaneous and articular syndrome (NOMID/CINCA).
The majority of MWS and FCAS cases are familial [64], while the majority of NOMID cases reported are sporadic [65, 66]. The latter is thought to be due to the severity of the NOMID phenotype with death prior to reproductive age. Among approximately 130 mutations identified in NLRP3/CIAS1, greater than 90 % have been associated with exon 3 [38, 64]. Mutations in NLRP3 result in inappropriate activation of the inflammasome and elevated IL-1 production. Approximately 50 % of NOMID patients have germline mutations in NLRP3 by Sanger sequencing while far fewer germline mutations have been found in MWS and FCAS. In those for whom germline mutations cannot be identified, approximately 70 % have somatic mosaicism identified through deep sequencing methods [67]. Given this information, diagnosis of CAPS is made on a clinical basis, with rapid response to trial with an IL-1 antagonist used for confirmation of clinical diagnosis. Genetic testing may be used for confirmation of diagnosis.
Common features in patients with CAPS include fever, leukocytosis, transient neutrophilic urticaria, conjunctivitis, arthralgias, and elevated inflammatory markers. The urticaria-like rash tends to be non-pruritic and presents as rose-colored flat macules or slightly raised plaques on the trunk and extremities that resolve within 24 hours. Lesional skin histopathology is characterized by neutrophilic infiltrate with interstitial and perivascular neutrophils and lymphocytes without dermal edema or vasculitis [68, 69]. Disease onset, severity, multiorgan involvement, morbidity and mortality differ between these three diseases.
FCAS
FCAS is the mildest of the 3 cryopyrinopathies. Patients usually present early in childhood, but presentations later in adulthood have been described. Febrile attacks are triggered by cold exposure, with development of fever and rash within 2 hours of exposure, peaking at 2–6 hours, and lasting 12–24 hours. Amyloid deposition is infrequent, and is found in only 2 % of patients [23, 70].
Muckle-Wells Syndrome
Muckle-Wells syndrome is characterized by intermediate severity. Unlike FCAS, disease manifestations are not associated with cold exposure and febrile attacks tend to occur with greater frequency, with the characteristic rash often noted in the early afternoon [71]. Ocular inflammation is characterized by inflammation of the conjunctiva, sclera, and anterior chamber. Patients can present with headache secondary to papilledema and aseptic meningitis. In the second or third decades of life, patients can develop sensorineural deafness due to damage of the Corti organ secondary to chronic ear inflammation. Sensorineural deafness occurs in approximately 75 % of untreated cases [72, 73]. AA amyloidosis is frequently seen in MWS, occurring in 25 % of untreated cases [74].
NOMID/CINCA
NOMID/CINCA is the most severe of the cryopyrinopathies. NOMID is characterized by chronic systemic multiorgan inflammation and persistently elevated inflammatory markers. Patients present within the first few weeks of life with persistent low-grade fevers, neutrophilic urticaria, painful arthropathy and elevated inflammatory markers (Fig. 40.3a). Aseptic meningitis and subsequent elevated intracranial pressure leads to irritability, headaches, nausea, vomiting and seizures. Although the aforementioned symptoms can wax and wane, inflammatory markers in patients with NOMID tend to remain persistently elevated [75].
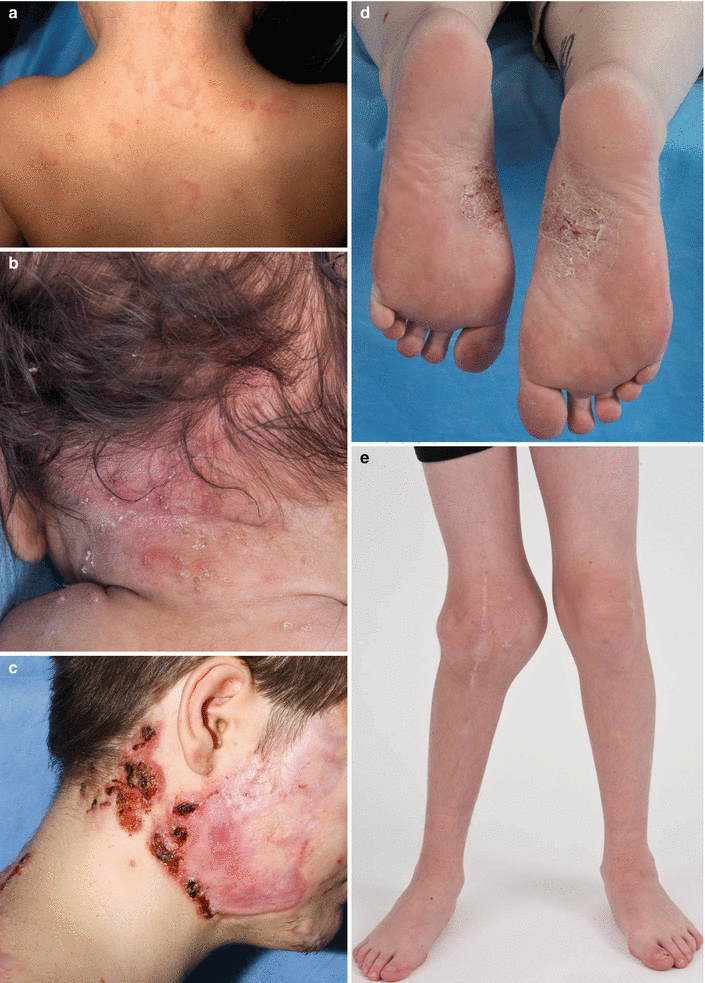
Fig. 40.3
Dermatologic manifestations of autoinflammatory diseases thought to be mediated by IL-1 pathways. (a) Urticaria-like plaques on the posterior neck and upper back of a child with neonatal onset multisystem inflammatory disease (NOMID). (b) Crop of pustules in the setting of background erythema on the posterior neck of an infant with deficiency of the IL-1 receptor antagonist (DIRA). (c) Pyoderma gangrenosum-like ulcers in the setting of acne and significant scarring involving the lateral and posterior neck and cheek of a young man with pyogenic arthritis, pyoderma gangrenosum and acne (PAPA). (d) Plantar pustulosis in a patient with SAPHO. (e) Hyperostosis of the right knee in the setting of CRMO
In untreated NOMID, significant end-organ damage is observed early in life [76]. Chronic aseptic meningitis leads to increased intracranial pressure, hydrocephalus and papilledema. Furthermore, chronic aseptic meningitis in combination with central nervous systemic inflammation can contribute to cognitive impairment. Chronic papilledema can result in optic nerve atrophy, leading to gradual vision loss in the third decade of life. Uveitis may also contribute to vision loss [75]. Sensorineural deafness due to chronic inflammation of the cochlea may develop within the first few years of life. Persistent joint inflammation resulting in premature and aberrant ossification, osteolytic lesions and cartilage hypertrophy can lead to permanent joint deformities in 50–70 % of untreated NOMID patients [65, 77–79].
All cryopyrinopathies respond rapidly and completely to IL-1 blockade [75, 80–86]. To date, three IL-1 antagonists – short-acting anakinra and the longer-acting rilonacept and canakinumab – have been FDA approved for the management of CAPS. IL-1 antagonists should be initiated early in life to prevent end-organ damage in these conditions and doses should be titrated to resolution of CNS and cochlear inflammation as continued benefit has been seen with prolonged use of these agents [4, 76, 79, 80, 83–85, 87, 88].
DIRA
Deficiency of the IL-1 receptor antagonist (DIRA) was first described in 2009 [89, 90]. Since that time, approximately 25 affected individuals in United States (including Puerto Rico), Canada, the Netherlands, and Brazil have been described [89–95]. Homozygous loss-of-function nonsense and missense mutations in the IL1RN gene, which encodes the IL-1 receptor antagonist (IL1RA), are responsible for the DIRA phenotype.
IL1RN is constitutively expressed in healthy human hosts and serves to inhibit proinflammatory IL-1-mediated signaling by competitively binding the IL-1 receptor. In DIRA, IL1RN mutations result in nonfunctional IL1RA, leading to unopposed IL-1 proinflammatory signaling and widespread systemic inflammation [96]. Because the IL-1 receptor is ubiquitously expressed, the disease manifestations of this condition are seen in multiple organ systems.
Affected individuals present at birth or within the first few weeks of life with fetal distress, pustulosis, oral ulcerations, joint swelling, and pain with movement (Fig. 40.3b). Premature birth has been observed. The pustular eruption of DIRA resembles pustular psoriasis, ranging from discrete crops of pustules to generalized pustulosis. The spectrum of bony changes seen includes epiphyseal ballooning of long bones, anterior rib-end widening, periosteal elevation of long bones and multifocal osteolytic lesions. High fevers are not a typical feature of DIRA. Leukocytosis and markedly elevated systemic inflammatory markers are found [89, 90]. Stomatitis, hepatosplenomegaly, respiratory insufficiency and thrombotic events have been less commonly observed. In untreated patients, mortality secondary to multiorgan failure in the setting of systemic inflammatory response syndrome (SIRS) and pulmonary hemosiderosis with progressive interstitial fibrosis has been reported.
Affected skin histopathology demonstrates epidermal acanthosis, hyperkeratosis and extensive epidermal and dermal neutrophilic infiltrate with pustule formation along hair follicles [89].
DIRA responds dramatically to IL-1 antagonist therapy, which effectively provides a recombinant replacement of the dysfunctional protein. Anakinra, a recombinant IL-1 receptor antagonist, leads to rapid improvement in skin manifestations, normalization of acute phase reactants and resolution of bony inflammation [89]. Given the severity of the disease and the availability of effective therapy, early disease recognition and prompt implementation of IL-1 blockade therapy prior to development of bony deformities, pulmonary sequelae and SIRS is critical.
DITRA
Deficiency of the IL-36 receptor antagonist (DITRA) was first described in 2011 in familial and sporadic cases of pustular psoriasis in 9 Tunisian families [97] and 3 unrelated English patients [98]. DITRA is an autosomal recessive condition caused by homozygous and compound heterozygous inactivating mutations in the IL36RN gene, which encodes the IL-36 receptor antagonist protein (IL36RA). Following the initial description of DITRA, IL36RN mutations have also been reported in individuals of varied ancestry, including European [99], Chinese [99], Japanese [100] and Lebanese [101], suggesting that these mutations may be found in other populations.
IL-36 is an IL-1 family cytokine that binds to the IL-36 receptor, enabling the recruitment of the IL-1 receptor accessory protein and subsequent signal transduction involving nuclear factor kappa-light chain-enhancer of activated B cells (NFkB) and mitogen-activated protein (MAP) kinases. The IL-36 receptor antagonist is encoded on chromosome 2 and competitively binds the IL-36 receptor, thereby providing negative feedback to IL-36 signaling [97, 98]. In DITRA, mutations in the IL36RN gene result in dysfunctional IL36RA protein, leading to unopposed proinflammatory signaling through the IL-36 receptor. Deficiency of the IL-36 receptor antagonist leads to an exaggerated inflammatory response by a mechanism analogous to that observed with dysfunctional IL-1 receptor antagonist protein in patients with DIRA. Unlike the IL-1 receptor in DIRA expression of the IL-36 receptor is limited to epithelial surfaces that have contact with the outside environment [98], thus limiting the disease phenotype predominantly to mucocutaneous surfaces.
The age of onset of DITRA is quite variable, ranging from onset in infancy through adulthood. Two cases of onset during pregnancy have also been reported [97]. Clinical characteristics include recurrent and sudden onset pustular eruptions on a background of erythematous plaques, with associated fevers, neutrophilia, leukocytosis and elevated inflammatory markers. While clinical features of DITRA are confined to the mucocutaneous surfaces, the spectrum of cutaneous involvement can be quite varied. While many reported cases demonstrate generalized pustulosis, patients have also been reported to have limited involvement including acrodermatitis continua of Hallopeau, palmoplantar pustulosis and migratory glossitis [97, 98], suggesting that the same gene may be responsible for a spectrum of phenotypes. Patients may have a history arthropathy, cholangitis, or plaque psoriasis [97]. Skin histopathology demonstrates epidermal acanthosis with elongation of rete ridges, parakeratosis and spongiform pustules [97].
Targeted therapeutic options for DITRA do not exist. The majority of reported DITRA patients have been managed with systemic and topical corticosteroids and systemic retinoids with variable success. Methotrexate and biologic agents, including adalimumab, infliximab and etanercept, have also been utilized with variable success [97, 98]. Recent reports of successful management of DITRA patients with IL-1 antagonists suggest that IL-1 might play an important role in disease pathogenesis [102, 103].
PAPA
Pyogenic arthritis, pyoderma gangrenosum and acne (PAPA) was first coined in 1997 [104]. It is an autosomal dominant disease with incomplete penetrance. Disease-causing mutations in PAPA syndrome are due to gain of function mutations in the proline-serine-threonine-phosphatase-interacting-protein-1 (PSTPIP1) gene, also known as CD2BP1.
PSTPIP1 is a cytoskeletal protein that interacts with PEST-type protein tyrosine phosphatases (PEST-PTPs), Wiskott-Aldrich syndrome protein (WASP), and pyrin. While the pathogenesis of PAPA syndrome is not completely understood, mutations in PSTPIP1 are thought to decrease the interaction of PSTPIP1 with PEST-type proteins, thereby increasing phosphorylation of PSTPIP1 and increasing PSTPIP1 interaction with pyrin, ultimately leading to unopposed IL-1 proinflammatory signaling [105].
PAPA patients typically present within the first decade of life with aseptic monoarthritis of the large joints such as knees, elbows and ankles. Persistent untreated chronic inflammation can lead to joint destruction. Cutaneous features of PAPA include acne and pyoderma gangrenosum. Although pyoderma gangrenosum lesions can present early in childhood, cystic acne tends to manifest at puberty (Fig. 40.3c). Hidradenitis suppurativa, psoriasis and rosacea have also been described [106–108]. Given the incomplete penetrance of this disease, a spectrum of disease severity is seen with PAPA, ranging from mild acne to explosive acne fulminans, pyoderma gangrenosum, and debilitating erosive joint disease. Pathergy is a prominent feature of PAPA.
Laboratory studies reveal persistently elevated systemic inflammatory markers and leukocytosis but are otherwise nonspecific. Histopathology of cystic acne lesions is similar to that seen with cystic acne in other settings: distended follicles with cystic spaces and follicular openings filled with keratinaceous debris and numerous bacteria. Ruptured cystic contents induce a brisk perifollicular neutrophilic inflammatory infiltrate. The histopathology of pyoderma gangrenosum is similar to that of pyoderma gangrenosum in other settings. Early lesions are characterized by a neutrophilic vascular infiltrate. Actively progressing lesions demonstrate neutrophilic infiltrates with leukocytoclasia. Pyoderma gangrenosum ulcers demonstrate marked tissue necrosis with surrounding mononuclear cell infiltrates. The synovial fluid from affected joints is characterized by sterile neutrophil-predominant infiltrate [108].
Management of PAPA with biologic agents has been met with mixed results. TNFα antagonists appear to be more effective for cutaneous manifestations while IL-1 antagonists seem to be more effective for managing articular disease manifestations. The reason for this is not known. Systemic and intra-articular corticosteroids have been used for articular disease, but steroid use can exacerbate acne so should be used with caution. Severe acne in PAPA patients can be managed with tetracycline antibiotics or isotretinoin. Effective management of PAPA syndrome is often a challenge and patients may require multiple systemic agents, including multiple biologic agents [109]. Patients should be closely monitored for infection when on multiple immunosuppressive agents.
SAPHO/CRMO
Although reports of acne and osteoarticular manifestations date back to the 1960s, the unifying term Synovitis Acne Pustulosis Hyperostosis Osteitis (SAPHO) was first described in 1987 [110, 111]. SAPHO is a rare disease, with a prevalence of fewer than 4 in 10,000 [112] and a female predilection [113–116]. This condition usually presents in childhood or young adulthood. Chronic Recurrent Multifocal Osteomyelitis (CRMO) was first described in 1972 and predominates in children [117]. It is unclear if SAPHO and CRMO are distinct diseases – many consider them to be the same entity. The genetic etiology of SAPHO/CRMO is unknown; however, a CRMO murine model does exist caused by homozygous mutations in the murine gene pstpip2 [118].
The etiology of SAPHO is poorly understood. A number of familial cases have been reported, suggesting a genetic component in disease pathogenesis [119–123]. In addition, SAPHO shares some clinical features with several monogenic autoinflammatory diseases, including DIRA and PAPA, in which pro-inflammatory IL-1 pathways are thought to play a critical role. The efficacy of IL-1 blockade in these other diseases suggests that IL-1 pathways may play an important role in SAPHO pathogenesis. Propionibacterium acnes, a skin saprophyte, has been isolated in bone biopsies of affected bone lesions [124–126], suggesting a possible role as an opportunistic pathogen that activates innate and T-cell immunity through increased complement, IL-1 [127], IL-8, and TNFα levels [128, 129].
Cutaneous manifestations of SAPHO and CRMO may present concomitantly with, prior to, or after the onset of osteoarticular disease. Approximately 85 % of adults with SAPHO develop cutaneous manifestations of disease while only 30 % of children with CRMO manifest cutaneous disease [130]. Cutaneous features are predominantly neutrophilic in etiology, with palmoplantar pustulosis predominating in 60 % of affected individuals, followed by acne seen in 25 % of patients [115] (Fig. 40.3d). Other reported cutaneous disease manifestations include hidradenitis suppurativa, dissecting cellulitis of the scalp, pilonidal cyst/sinus, generalized pustular psoriasis, psoriasis vulgaris, subcorneal pustular dermatosis, erythema nodosum, and more rarely, pyoderma gangrenosum or Sweet’s Syndrome [113, 131–134].
Osteoarticular manifestations are the hallmark of SAPHO/CRMO and are required for diagnosis. The osteoarthropathy is characterized most prominently by osteitis and hyperostosis. In adults, anterior chest wall osteitis is most common, affecting 65–90 % of individuals. The next most commonly affected site is the spine, affecting approximately 30 % of individuals, with the thoracic spine most frequently involved. Sacroiliitis develops in approximately 52 % of affected individuals. In adults, appendicular skeleton abnormalities are rarely seen, with long bone involvement in 5–10 %, and mandibular involvement reported in 1–10 % [135, 136]. In contrast, long bones are commonly involved in children, with distal and proximal tibia being most commonly involved, followed by proximal and distal femur [137, 138] (Fig. 40.3e). Affected individuals may have arthritis at joints adjacent to bony lesions. In adults, distant synovitis can also be seen.
Systemic features of SAPHO/CRMO include fevers and malaise. Elevated inflammatory markers can sometimes be seen. Inflammatory bowel disease, most often Crohn disease, has been reported in 10 % of patients with SAPHO syndrome [115].
First-line therapy for SAPHO/CRMO-related bony disease includes NSAIDs [139]. In the setting of NSAID failure, bisphosphonates [140–143] or methotrexate [139] should be considered for management of bone inflammation. Bisphosphonates are quite effective for the management of osteoarticular inflammation, but must be used with caution in females of childbearing age given the potential risks of fetal skeletal defects. In addition to efficacy in the management of bone inflammation, methotrexate has also demonstrated efficacy for skin manifestations. TNF-α antagonists [144] and IL-1 antagonists [145, 146] have also been shown to be highly effective for the management of refractory cases of SAPHO/CRMO skin and bone lesions in case reports and case series, however neither have been systematically studied and neither are FDA-approved for this indication.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


