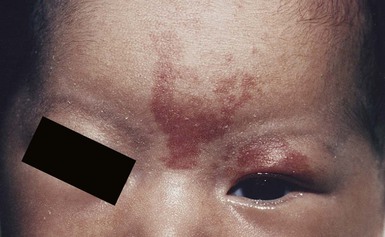
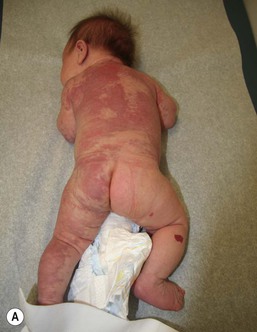
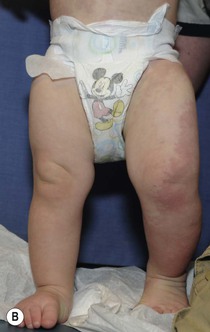
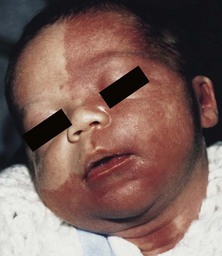
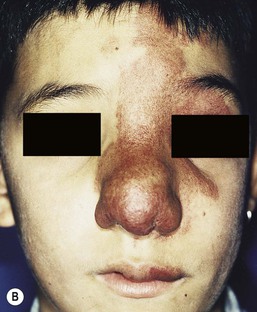
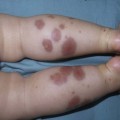
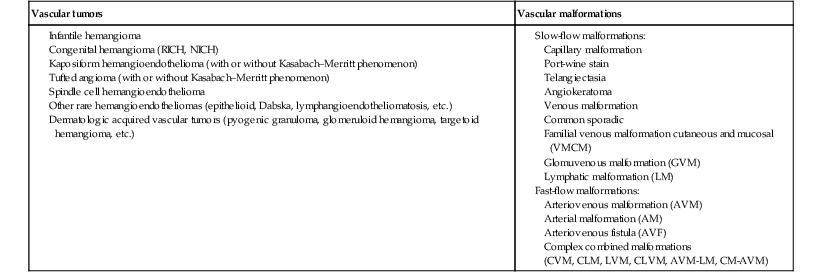
Sheilagh M. Maguiness, Maria C. Garzon In 1982, Mulliken and Glowacki1 proposed a biologic classification of vascular birthmarks that has become widely accepted. It was modified slightly in 1996 by the International Society for the Study of Vascular Anomalies (ISSVA).2 Two major groups of vascular birthmarks are recognized: vascular malformations, which are composed of dysplastic, malformed vessels; and vascular tumors, which demonstrate cellular hyperplasia. The distinction between malformations and tumors is emphasized by their varying histologic appearance, cellular markers, and natural history (see Chapter 21 for a full review of vascular tumors).3,4 The ISSVA classification of vascular anomalies allows for more precise diagnoses and helps avoid the perpetuation of out-dated terminology such as ‘cavernous hemangioma,’ which has been previously used to describe both tumors and malformations (Table 22.1).3 TABLE 22.1 ISSVA Classification of vascular anomalies (Mulliken and Glowacki 1982; The International Society for the Study of Vascular Anomalies, ISSVA 1996, 2003) Adapted from Enjolras O, Wassef M, Chapot R. Color atlas of vascular tumors and vascular malformations. New York, NY: Cambridge University Press; 2007. Vascular malformations are subcategorized according to flow characteristics and predominant anomalous channels. They can occur alone or in combination: slow-flow malformations (capillary, C; venous, V; lymphatic, L) or fast-flow malformations (arteriovenous malformation, AVM; and arteriovenous fistula, AVF). Vascular malformations are often localized and circumscribed lesions, but can also present in a segmental/regional pattern or in a multifocal, disseminated form. Vascular malformations can also be observed in the setting of various syndromes, many of which arise due to known genetic mutations. In several cases, the discovery of the genetic mutation has advanced our understanding of the pathogenesis of the underlying conditions. A number of complex and/or combined vascular malformations also exist: CVM, CLVM, LVM, AVM, CAVM, CLAVM, and so forth, with some of them known by eponyms. In this chapter, we discuss a variety of these sometimes eponymous syndromes associated with vascular malformations. We have grouped them according to their predominant malformation but recognize these categories are not always rigid and significant overlap exists. A salmon patch is a capillary malformation (CM), also known as an ‘angel kiss’ when located on the forehead or eyelids, and ‘stork bite’ when located on the nape of the neck. It is present in nearly half of all newborns and affects males and females equally. It has a characteristic predilection for the midline, with the most common locations being the nape, upper glabella, nose, and upper lip (Fig. 22.1). The occiput and lower back may also be affected.5 If a salmon patch involves the upper eyelid and does not occur concomitantly with the classic V-shaped patch in the middle of the forehead, it may be difficult to differentiate from a partial V1 port-wine stain or a hemangioma precursor. Occasionally, widespread involvement of nevus simplex at many different sites can exist – the term ‘nevus simplex complex’ has recently been proposed to describe such cases in an attempt to prevent confusion with port-wine stains.6 Nevus simplex usually disappears within 1 or 2 years, but some persist, particularly those at the nape.7 A nevus simplex localized to the midline sacral region has sometimes been referred to as a ‘butterfly-shaped mark’. Involvement at this site is most often seen in infants with multiple salmon patches elsewhere. In most cases, these are benign and not associated with underlying spinal dysraphism. However, prospective studies with MR imaging are lacking, so it is not possible to exclude this risk completely. In the absence of any other concerning signs, a nevus simplex in this location is usually innocent. Although nevus simplex usually do not have underlying associations, rarely they are a manifestation of certain syndromes, such as Beckwith–Wiedemann, macrocephaly–capillary malformation (MCM/MCAP), or Nova syndromes (see below). The terms port-wine stain (PWS) and capillary malformation (CM) are used interchangeably. CMs are almost always evident at birth and found in up to 0.3% of newborns.8 They are mostly sporadic in origin. Histopathologically, they are comprised of normal capillaries within the superficial dermis without evidence of proliferation. CMs can be observed in the setting of several different syndromes discussed later in this chapter. The etiology of CM is not fully understood, however recent reports of mutations in GNAQ in both syndromic and non-syndromic CMs point to an underlying genetic basis.9 Another familial form of CM occurring with AVM has been reported in the CM-AVM syndrome. PWS are pink or red patches that typically grow proportionately with the child’s somatic growth, and persist throughout the patient’s life. They consist of ectatic dermal capillaries and may occur anywhere on the body (Fig. 22.2A). PWS may appear to lighten over the first 3–6 months of life, but this should not be taken as a sign of resolution. Rather, this physiologic lightening is due to the decrease in blood hemoglobin concentration corresponding to the change from fetal to adult type hemoglobin (Fig 22.2B).10 Capillary malformations can occur anywhere on the body. They may or may not be associated with overgrowth of the affected area (Fig. 22.2). Port-wine stains on the face tend to follow a dermatomal pattern, respecting the trigeminal nerve V1-V3 distribution or comprising multiple dermatomes (Fig. 22.3). The natural history of port-wine stains over a lifetime is often one of gradual darkening from pink-red to a crimson or deep purple hue. Skin thickening and soft tissue and/or bony hypertrophy may also develop. Eczematous changes can occur in PWS and salmon patches, either with or without treatment. Nodular vascular lesions (pyogenic granulomas) may appear within PWS during childhood or adult life and may require surgical intervention.11 The association of PWS with pigmentary anomalies such as extensive Mongolian spots, nevus spilus, or nevoid hyperpigmentation is a feature of phakomatosis pigmentovascularis (see further discussion of this entity below, Fig. 22.4, Table 22.2). Progressive soft tissue and bony overgrowth may occur during childhood, especially with V2 PWS, and subtle changes are sometimes noted even in the neonatal period.12 Asymmetric maxillary hypertrophy associated with distortion of the facial features requires multidisciplinary management with orthodontic follow-up and treatment. Some patients require procedures to correct skeletal overgrowth in late childhood. Gingival hypertrophy may also develop. Port-wine stains are also a manifestation of several different syndromes with other extracutaneous features discussed later in the chapter (see ‘Syndromes associated with vascular malformations’, below). The gradual thickening and nodularity of PWS provide a medical rationale for treatment during infancy and childhood.11 The flashlamp-pumped pulsed dye laser (PDL) is the gold standard treatment for PWS and poses a very low risk of scarring, even in young infants. It has been used for longer than 15 years and advances such as the development of a cooling system have improved efficacy. Most PDL units have a wavelength of 595 nm. Settings of 0.45–1.5 ms pulse duration, 6–10 J/cm2 fluences, and 7–10 mm spot size along with the coolant system (dynamic cooling device) are used in the majority of cases. Although only 15–20% of PWS clear completely with PDL, the majority of treated lesions lighten significantly.13,14 Response to laser treatment varies by area and skin type: outcomes are better on the face and neck, albeit with less improvement in V2 than other facial sites, and are also better in patients with type I–II skin.14 The extremities do not respond as well.13–15 Controversy exists regarding the age at which treatment should begin. Some authors have noted a better therapeutic response with fewer treatments when treatment was initiated in early infancy,16 but others have found no difference.17 Treating earlier (in infancy or early childhood) can be helpful in reducing stigmatization and may help prevent thickening of the skin.18 Treating early in infancy may enable the physician to treat with topical anesthesia, as small infants can be swaddled and held during a brief treatment and local anesthetic can be applied prior to the procedure. In older children with large stains, general anesthesia may be required. In most cases, re-darkening occurs with time and touch-up treatments are often needed in later childhood or teen years.19 In treatment-resistant CM, other laser modalities have recently been studied in an attempt to decrease thickness, color, and nodularity. These include the long-pulsed neodymium : yttrium-aluminum-garnet (Nd : YAG) laser, the combined PDL and Nd : YAG laser, and the alexandrite laser.20–24 Other novel therapies show some initial promise, including intense pulsed light (IPL), photodynamic therapy (PDT), and application of topical anti-angiogenic factors (imiquimod and rapamycin) in conjunction with laser ablation.25–35 In cases where there is soft tissue or bony overgrowth, or in syndromic CM, a multidisciplinary approach to management is often necessary. Patients may require orthodontic work, maxillofacial reconstruction or soft tissue debulking. Ideally, follow-up in a multidisciplinary vascular anomalies center is beneficial. Though not true CMs, telangiectasias can present in infancy or early childhood and often persist. They are usually composed of small, punctate linear vessels distributed in either a segmental, unilateral nevoid, or diffuse pattern. A variety of telangiectatic skin lesions have been described. Most are absent at birth, often developing in childhood. A pale halo of vasoconstriction may surround small telangiectasias. Toddlers and young children sometimes develop so-called spider angiomas comprised of a brightly erythematous central punctum with radiating ‘spider-like’ telangiectasias. They are usually located on sun-exposed areas. Risk factors include fair skin and a history of minor skin injury at the site. These may disappear spontaneously over years, or persist. Fine telangiectasias are occasionally present on the cheeks of normal young children, but extensive telangiectasias in the photo-distribution should prompt consideration of conditions with photosensitivity such as Rothmund–Thomson syndrome. Persistent telangiectasias can also be seen as a sequela of neonatal lupus. The facial telangiectasias associated with hereditary hemorrhagic telangiectasia (HHT) and ataxia–telangiectasia usually present later in life. Venous malformations (VM) are slow-flow vascular malformations that are usually evident at birth. They may involve skin, mucosa, subcutaneous tissues, muscle and rarely bone. They are composed of ill-defined venous channels with irregularly attenuated walls, focally lacking smooth muscle cells, permeating the skin and adnexal structures. Glomuvenous malformation (GVM) is a subtype of VM composed of anomalous venous channels lined by glomus cells, which are actin positive and do not lack smooth muscle. Glomus cells have a characteristic appearance on histopathology. VMs are sporadic in the majority of cases, but, familial cases of both VM and (more often) GVM occur. The genetic basis for familial VMs (VMCM syndrome, see below) and GVM are now known. The mutated gene in familial VMCM syndrome maps to chromosome 9p17; and is an activating mutation in the kinase domain of the receptor tyrosine kinase Tie2.36 Sporadic Tie2 mutations are also found in 50% of sporadic VMs.37 GVM can be either a sporadic or an autosomal dominant condition, linked to mutations in the glomulin gene (mapping to 1p21–p22). Unlike VMs, far more cases of GVM are familial (approx. 64%).38,39 At birth, the majority of VM are subtle, bluish, ill-defined, compressible plaques or nodules. Rarely, a bulky venous mass is observed in the neonatal period (Fig. 22.5). VMs may be diffuse or localized. Affected skin and mucous membranes are typically blue with normal skin temperature, without a thrill or bruit. Lesions swell when dependent or with crying. In many cases, the blue-hued cutaneous component heralds involvement of deeper structures. Local venous thromboses can lead to the formation of phleboliths, which can be tender when palpated and are visible on radiographs as round calcifications. Glomuvenous malformations (GVM; formerly called ‘glomangioma’) may be solitary or multiple, and may be localized or involve a larger territory of skin (Fig. 22.6). They are often bluish to purple, cobblestoned or plaque-like in appearance. During childhood, they typically acquire a deeper blue hue and thicken, and become tender when palpated. Congenital plaque-like GVMs are usually pink at birth, with noticeable thickening and change in color to blue-purple during childhood. These plaque-like GVMs may arise sporadically, or occur as a manifestation of autosomal dominant GVM. Other family members may have smaller, blue vascular lesions scattered over the skin, increasing in number with age.38 In addition to skin and mucosal involvement, VM can also involve deeper soft tissues, muscles, joints, and in severe cases, visceral sites such as the abdomen and pelvis. A majority of patients with extensive VMs develop a chronic localized intravascular coagulopathy (LIC), which is distinct from Kasabach–Merritt phenomenon (KMP). VM associated LIC can result in either thrombosis (with pain and phlebolith formation) or bleeding, and persists throughout life. It differs from KMP because the primary process is one of ongoing clotting, with consumption of clotting factors, low fibrinogen and elevated d-dimers, but without the marked thrombocytopenia of KMP.40,41 Large and intramuscular VMs are more likely to have associated VM-LIC. Pain is a common complaint either due to phlebolith formation or often in larger extremity lesions.42 ‘Blue rubber bleb nevus’ syndrome, the association of multiple cutaneous venous malformations with gastrointestinal and other internal lesions, is discussed later in this chapter. Some patients with VM experience worsening of symptoms during puberty or in pregnancy. In contrast to VMs, GVMs are typically localized to the skin and soft tissues without mucosal or intramuscular involvement. GVMs are not associated with LIC. The diagnosis is usually established on the basis of clinical features. Imaging modalities including ultrasonography, Doppler, MRI, and CT scans are useful for evaluating the extent of involvement.43 The finding of associated LIC and specifically elevated d-dimer levels, can help confirm the diagnosis of VM in uncertain cases.44 Apart from helping to confirm the diagnosis, the decision to image early in infancy depends on whether functional problems are present or early treatments are planned. Many VMs, however, especially larger ones, eventually do require imaging studies, and MRI with contrast is the best study to delineate disease. A number of radiologic classification schema have been proposed and may predict response to treatment, with smaller and more localized lesions being more amenable to sclerotherapy.45 In individuals with craniofacial VMs, it is advisable to image the brain: developmental venous anomalies (DVA) in the brain are more common in patients with craniofacial VMs than in the general population (25% vs 0.5%).46 DVAs are uncommon trajectories of the brain’s venous drainage and pose little risk of cerebral hemorrhage, but documenting their presence can help avoid misdiagnosis of a more worrisome condition later in life. Histopathology can also help to confirm the diagnosis when uncertainty is present and/or to distinguish between VM and GVM. Extensive VMs in a leg or an arm and adjacent trunk must be differentiated from Klippel–Trenaunay and other overgrowth syndromes, as management and prognosis is variable (see Table 22.4). Sinus pericranii should be considered in the differential diagnosis of a VM located on the central forehead. This presents as a bluish, nonpulsating mass that is usually congenital and quickly expands when the patient puts their head in a dependent position or with crying. Sinus pericranii represents a direct communication between superficial veins and intracranial venous sinuses through a bony defect. It is best imaged using CT with bone windows. Venous malformations tend to enlarge and become more symptomatic with time. Their clinical course and complications depend on anatomic location, with differing problems in the craniofacial area, trunk, and limbs.41 The cheek, lip and tongue are common locations for craniofacial VMs, and there is sometimes deep extension to the temporal and orbital areas. Swelling is noted with dependency and activity. Over time, the VM may progressively distort the facial features and mold the underlying developing bones, which can result in deformities such as an open bite or enlargement of the orbit. Extensive retropharyngeal involvement can result in obstructive sleep apnea, occasionally even in young children. VMs located on the trunk and limbs may involve skin, skeletal muscles, joints, and bones, often causing pain. During infancy and early childhood, the skin component of the VM expands and becomes deep blue. However, the deeper component may remain undiagnosed until it becomes symptomatic. Swelling, functional impairment, and limited joint motion occur when the child becomes older and more active, especially if playing sports. VM-LIC associated with large and intramuscular VM can manifest as early as the neonatal period. Due to the heterogeneity of VMs and the clinical and radiologic diagnostic overlap with other vascular anomalies, multidisciplinary management is recommended. VMs are reported to comprise up to 40% of referrals to vascular anomalies centers.47 VMs can be treated with many different modalities including sclerotherapy, excision, endovenous laser ablation, cutaneous laser ablation or a combination of these modalities. Sclerotherapy has emerged as the mainstay of treatment for VMs, followed by surgical excision. The decision regarding when to treat is usually based on whether there is significant functional impairment or disfigurement. Sclerotherapy involves the introduction of an endothelial-cell cidal sclerosant into the vascular spaces of the malformation.48 Several different sclerosants can be used, including ethanol or sodium tetradecyl sulfate. Complications of sclerotherapy include tissue necrosis, nerve palsies, hemoglobinuria and oliguria.49 Surgical management can be most helpful in small or localized lesions. A combination of sclerotherapy followed by surgical debulking is often used in large or complex cases. In craniofacial lesions, therapy is aimed at preventing distortion of facial anatomy, limiting bone deformity, open bite or shift of the dental midline, lip expansion, and displacement of the lip commissures.50,51 MRI and ultrasound features may be especially helpful in determining optimal management.51 Multiple treatments are often required over years. Laser or radiofrequency ablation are occasionally helpful. Conservative therapy with compression should be initiated early on and is even encouraged from infancy in diffuse extremity VMs. Compression increases comfort, limits swelling, and improves coagulopathy. Compression is less useful for intramuscular VMs and may exacerbate pain. Evaluation of large VMs includes assessment for, and possibly treatment of, VM-LIC. d-dimer levels should be obtained periodically to assess the extent of coagulopathy. Treatment with low-dose aspirin can be a helpful adjunct to conservative therapy in some cases. Low molecular weight heparin (LMWH) has been found to be useful in patients with chronic VM-LIC during episodes of exacerbation (thrombosis, pain) or before and after planned procedures such as sclerotherapy or surgical excision, to prevent thrombosis and/or control intraoperative hemorrhage.52 In patients with a history of thromboembolic events or in those with high risk, large and/or combined malformations, placement of an inferior or superior vena cava filter may help prevent pulmonary embolus.53 Lymphatic malformations (LMs) are slow-flow vascular anomalies known in much of the literature as ‘lymphangioma’. They can be macrocystic, microcystic, combined or very rarely generalized. LMs arise in a sporadic manner and can also be syndromic. The etiology of LMs is still unknown. LMs arise in a sporadic manner and no associated genetic basis has yet been discovered. This may suggest that a genetic cause is likely related to somatic mutations, which, if occurring within the germline, would be incompatible with life.54 This is in contrast to primary lymphedema, where several genetic mutations leading to specific lymphedema syndromes have been discovered and have improved our understanding of the molecular pathways involved in lymphangiogenesis.55 In primary lymphedema, defective lymphatic drainage leads to accumulation of lymphatic fluid in the interstitial space (Fig. 22.7). Several forms of lymphedema are known, including familial congenital lymphedema, Nonne–Milroy syndrome, Meige disease, and Hennekam syndrome, summarized in Table 22.3. TABLE 22.3 Hereditary lymphedema syndromes Macrocystic LMs are usually visible at birth and are commonly diagnosed by prenatal ultrasound investigation. They occur more commonly in the neck and axilla, where they are often referred to as cystic hygroma (Fig. 22.8).56 The detection in utero of some huge LMs of the neck, axilla, and thoracic area may lead to a discussion about terminating the pregnancy, as the prognosis is poor. Microcystic LMs can infiltrate diffusely throughout the dermis. Clear or hemorrhagic vesicles (so-called ‘lymphangioma circumscriptum’), which may intermittently leak lymphatic fluid, may be visible on the surface of the lesion. Severe combined LMs may also occur on the trunk and limbs.57 Cervicofacial LMs involving the tongue and floor of the mouth will interfere with normal development of the jaw and create an open bite deformity. The most severe forms of combined micro/macrocystic LMs, which are more common on the head and neck, can cause life-threatening airway disease and other functional problems in the neonate (Fig. 22.9). Intraoral involvement also causes drainage of serosanguineous lymphatic fluid, aggressive caries, and loss of teeth. Involvement of the mandible with ensuing overgrowth is present in about 40% of these patients.58,59 Extra- and intraconal orbital LM occurs in association with eyelid LM; this uncommon location causes severe complications including disfigurement, bleeding, infection, proptosis, and visual loss.60 Severe cervicofacial LMs, usually the combined micro/macrocystic type, involving the hypopharynx and larynx, the tongue and the floor of the mouth, can cause airway and esophageal obstruction requiring nasogastric tube feeding and emergency tracheotomy in the newborn.61 Of 31 cases with such severe involvement, 58% required tracheostomy in infancy and one-third could not be decannulated.59 Visceral LMs, intrathoracic or abdominal, are less common, representing about one-tenth of cases. LMs are more susceptible to bacterial infections, and recurrent cellulitis can be a complication. The diagnosis is established either clinically or using CT, MRI, or ultrasound. Fluid aspiration and analysis may be helpful because a number of neonatal growths, including some malignant tumors, can present with large cyst-like swellings.62 Histologically, lymphatic vessels or cysts have thin walls and lumina appear empty. Several positive immunohistochemical markers of lymphatic vessels have been identified, including LYVE-1, VEGFR-3 and D2-40, and can help to differentiate LM from other vascular anomalies. Lymphatic malformations are benign and treatment is usually directed at managing complications or attempting to restore anatomy. Conservative measures include observation, prophylactic antibiotics when infection is a concern, and in the case of primary lymphedema, compression. Sclerotherapy has emerged as an important treatment for LMs, and can be done early, even in the neonatal period. A variety of sclerosing agents have been used, including doxycycline, ethanol, bleomycin, OK-432 and STS. Macrocystic LMs respond very well to sclerotherapy. Surgical resection is another therapeutic option for macrocystic LMs, as well as for microcystic and combined types, but recurrences and complications such as seroma or chronic lymphatic leakage may occur.58–60,63,64 Radiofrequency ablation can be helpful in the management of LMs, most commonly for the lips and tongue to help decrease chronic leakage and bleeding.65,66 A multidisciplinary approach is required for complex LMs.59 Arteriovenous malformations (AVMs) are fast-flow anomalies that most commonly arise in the head and neck area. Macular erythema mimicking a PWS, with increased local warmth, and subtle skin thickening can be clues to the diagnosis. Arteriovenous malformations arise due to errors in vascular development, which occur during embryogenesis. AVMs represent direct shunting from the arterial to the venous system due to the absence of an intervening capillary bed. Recently, the genetic basis for familial types of AVM has been discovered. Mutations in RASA1 have been found in individuals with AVM of the extremity (Parkes Weber syndrome) and in CM AVM syndrome (see below for further discussion on Parkes Weber syndrome), vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies caused by RASA1 mutations.9 Other mutations associated with AVMs or AVM variants include endoglin, which gives rise to hereditary hemorrhagic telangiectasia syndrome (HHT) and PTEN as seen in high flow lesions of the PTEN hamartoma tumor syndrome (see below). Most commonly, AVMs present as a pink vascular stain on the skin, mimicking port-wine stain. They are often located on the head and neck, the nose and central face/lip are common locations as is the ear (Fig. 22.10). As mentioned above, lesions are warm and may have a thrill or a bruit. AVMs may develop pyogenic granulomas within them that bleed and may need to be excised or treated with laser ablation.67 Any patient who presents with small, multifocal lesions that resemble PWS but demonstrate high flow on Doppler examination should receive a workup for CM AVM syndrome (see below). Lesions may become thicker, more nodular and darker in color with time. AVMs often progress during puberty or with trauma. AVMs can be life-threatening lesions that worsen over time. Complications include disfigurement, pain, bony erosion, hemorrhage, and even death. Large AVMs may have associated high-output heart failure at birth or later in life, requiring intervention. Most AVMs can be diagnosed based on their natural history and physical examination characteristics. They may not be obvious at birth, most commonly lesions mimic port-wine stain, but unlike PWS, AVMs expand through childhood. In general, AVMs are warm, a bruit may be audible, and a thrill may be palpable. An actual vascular mass with tense draining veins, a thrill, and a bruit is uncommon for AVM in a newborn, and may represent a congenital hemangioma (see Chapter 21
Vascular Malformations
Introduction to vascular malformations
Vascular tumors
Vascular malformations
Capillary malformations
Nevus simplex (salmon patch, fading capillary stain)
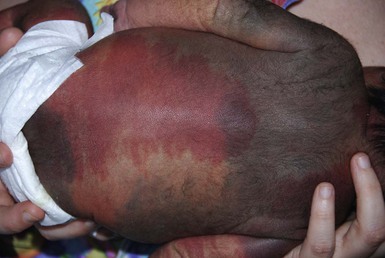
Capillary malformations
Pathogenesis
Cutaneous findings
Extracutaneous findings
Management
Telangiectasia
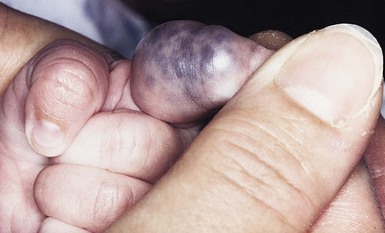
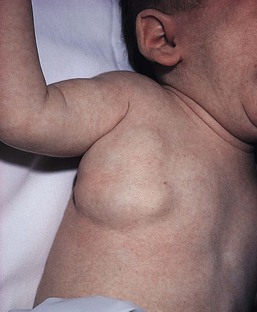
Venous malformations
Pathogenesis
Cutaneous findings
Extracutaneous findings
Diagnosis
Differential diagnosis
Course
Management
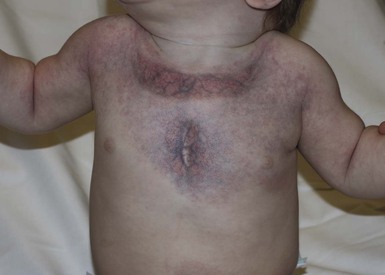
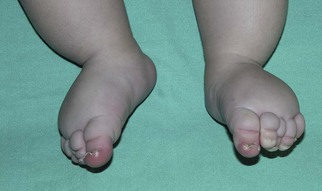
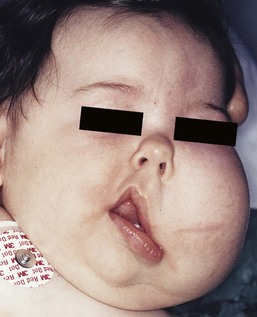
Lymphatic malformations
Pathogenesis
Syndrome
Genetic basis
Clinical features
Primary congenital lymphedema (Nonne–Milroy disease)
Autosomal dominant
Mutations in VEGFR3
Congenital lymphedema of lower limbs, mainly below knees. Slow to progress. Can be associated with hydrocele, enlarged leg veins and recurrent cellulitis
Pubertal onset lymphedema (Meige disease, lymphedema distichiasis syndrome)
Mutations in FOXC2
Pubertal onset, most common type of primary lymphedema. Can be associated with distichiasis, ptosis, yellow nails, syndactyly, cleft palate and cardiac septal defects
Hypotrichosis–Lymphedema–Telangiectasia syndrome
Autosomal dominant and recessive forms reported mutations in SOX18
Lymphedema, sparse hair, cutaneous telangiectasias
Hennekam syndrome (generalized lymphatic dysplasia, GLD)
Autosomal recessive
Mutations in CCBE1 in some cases
Lymphedema, intestinal lymphangiectasia, protein-losing enteropathy, developmental delay, multiorgan involvement
Cutaneous findings
Extracutaneous findings
Diagnosis
Treatment
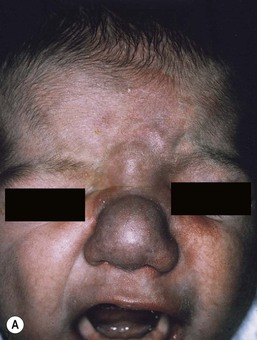
Arteriovenous malformations
Pathogenesis
Cutaneous findings
Extracutaneous findings
Diagnosis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Vascular Malformations
22