TLR
Disease
Comments
1
Tuberculoid
TLR1 favors Th1 phenotype [11]
Leprosy
Psoriasis
TLR1 expression increased in keratinocytes [13]
Lyme disease
Syphilis
Increased neurosyphilis risk in TLR1 polymorphisms [16]
2
Acne vulgaris
P. acnes stimulates TLR2 and causes hypercornification of sebaceous glands [17]
Atopic dermatitis
Psoriasis
Increased TLR2 expression in keratinocytes [13]
Staphylococcus aureus infection
Leprematous leprosy
Associated with Arg677Trp mutation in Korean population [33]
Arg677Trp mutation: decreased cytokine production [34]
Syphilis
Lipoproteins stimulate TLR2 [35]
Increased neurosyphilis risk in TLR2 polymorphisms [16]
Lyme disease
Outer surface proteins stimulate TLR2 [36]
Patients with Arg753Gln mutation secreted less proinflammatory cytokines [37]
Candidiasis
HSV
TLR2−/− animals are more susceptible to HSV encephalitis [42]
3
Psoriasis
Mutation in AP1S3, gene required for TLR3 trafficking, associated with pustular psoriasis [43]
HSV
TLR3−/− astrocytes fail to produce type I IFN [44]
Humans with TLR3 deficiencies are more susceptible to HSV encephalitis [10]
4
Acne vulgaris
P. acnes LPS stimulates TLR4 [45]
Allergic contact dermatitis
Psoriasis
Syphilis
Lipoproteins stimulate TLR4 [35]
Candidiasis
Important for neutrophil recruitment [51]
UV exposure
TLR4 hyporesponsiveness leads to impaired TNFα production [52]
TLR4-MyD88 axis deficiencies led to increased cell survival and upregulation of necroptic markers [53]
TLR4 deficiency led to increased nucleotide excision repair [54]
6
Syphilis
Increased neurosyphilis risk in TLR6 polymorphisms [16]
7
Psoriasis
Systematic lupus erythematous (SLE)
pDCs bind self nucleic acids to stimulate IFN production via TLR7 and 9 [57]
Small nuclear RNA binds and activates TLR7 and 8 [58]
Gene duplications of TLR7 increases autoantibody production [59]
Chronic TLR7 and 9 stimulation leads to glucocorticoid resistance [60]
Melanoma
Mycosis fungoides
Imiquimod shown to have clinical responses [65]
UV exposure
Imiquimod enhances DNA repair and decreased DNA damage [66]
8
SLE
Small nuclear RNA binds and activates TLR7 and 8 [58]
9
Atopic dermatitis
Polymorphisms associated with disease [67]
Psoriasis
DNA complex with LL-37 stimulates TLR9 to drive IFNα-mediated inflammation [68]
SLE
pDCs bind self nucleic acids to stimulate IFN production via TLR7 and 9 [57]
Chronic TLR7 and 9 stimulation leads to glucocorticoid resistance [60]
Melanoma
Mycosis fungoides
Discovery of TLRs in Humans and Its Expanding Role in Immunity
After Janeway proposed the theory of pattern recognition, based on what was then known about other innate immune receptors, his group was in search for cell-surface receptors expressed on APCs that resulted in NF-κB activation [77]. Lemaitre et al. first identified the antifungal function of Drosophila Toll and demonstrated that it plays a key role in regulating antibacterial gene expression through the NF-κB-like signaling pathway [78]. This seminal discovery paved the path for the discovery of its human counterpart in which Janeway et al. [79] demonstrated that the mammalian Toll homolog induced expression of genes encoding B7 and cytokines that affect the adaptive immune response, providing confirmation for the theory of pattern recognition. Researchers began a fervent search for the ligand of human Toll (now known as TLR4). The first clue came when researchers found that C3H/HeJ mice were unresponsive to bacterial lipopolysaccharide (LPS) and mapped the genetic locus required for LPS responsiveness to TLR4 [80, 81]. Subsequent studies that attempted to clarify this ligand-receptor interaction proved to be difficult until the other protein in the receptor complex, MD2, was discovered [77, 82]. Since then, studies by many groups have identified multiple other members in the TLR family and elucidated many of their ligands [83]. For their efforts in discovering the toll receptors in Drosophila, Bruce Beutler and Jules Hoffmann won the Nobel Prize in Physiology or Medicine in 2011. TLRs are now the most well characterized PRRs and it is established that different TLR members recognize a variety of PAMPs. Up to 13 TLRs have been identified in mice but only 10 are present in humans as TLR11, 12 and 13 have been lost from the human genome [84]. In contrast, the C-terminal of TLR10 in mice is disrupted by a retrovirus insertion and is nonfunctional. For a detailed look at the history of TLRs, see Table 2.2.
Table 2.2
Historical timeline: discovery of Toll-like receptors
Discovery | |
|---|---|
1979 | Identification of the dorsal mutation [85] |
1984 | Characterization of toll mutation and other dorsoventral mutations |
1989 | Janeway proposes the theory of pattern recognition [2] |
1993 | Demonstration that NF-κB is required for Drosophila antimicrobial resistance 209 |
1996 | Drosophila Toll identified; found to be required for resistance to fungal infections [78] |
1997 | Human homologue of Drosophila Toll, signals activation of adaptive immunity [79] |
1998 | |
1999 | MD2 identified as coreceptor for TLR4-LPS interaction [82] |
2000 | TLR9 recognizes bacterial DNA [87] |
2000 | TLR2 can pair with TLR6 to recognize bacterial proteins [88] |
2000 | TLR2 can also associate with TLR1 [88] |
2001 | TLR3 mediates response to viral double-standed RNA [89] |
2001 | TLR5 detects flagellate protein in whiplike tails of bacteria [90] |
2004 | TLR8 (humans), TLR 7 (mice) recognize single-stranded RNA [91] |
2011 | Bruce Beutler and Jules Hoffmann awarded the Nobel Prize in Medicine for their role in the identification of TLRs |
As our understanding of TLRs has expanded in the past couple of decades, increasing evidence has indicated that TLRs are not limited to recognizing PAMPs but can also bind to signals released from damaged tissues, a notion first pioneered by Polly Matzinger who proposed the danger theory as an alternative to the mechanism of immunity initiation [92]. Non-pathogen associated material that leads to tissue injury and other endogenous ligands released during cellular injury such as chromatin bound high mobility group 1 and heat shock proteins also bind and activate TLR signaling [93–97]. Thus, in addition to being the first line of defense against pathogens, TLRs also survey the expression of danger-associated molecular patterns (DAMPs) seen in tissue injury (Fig. 2.1). TLR activation by DAMPs results in sterile inflammation that may play a role in chronic skin diseases such as psoriasis (Fig. 2.2) [99]. For a detailed look at PAMPs and DAMPs that activate specific TLRs, please see Table 2.3.
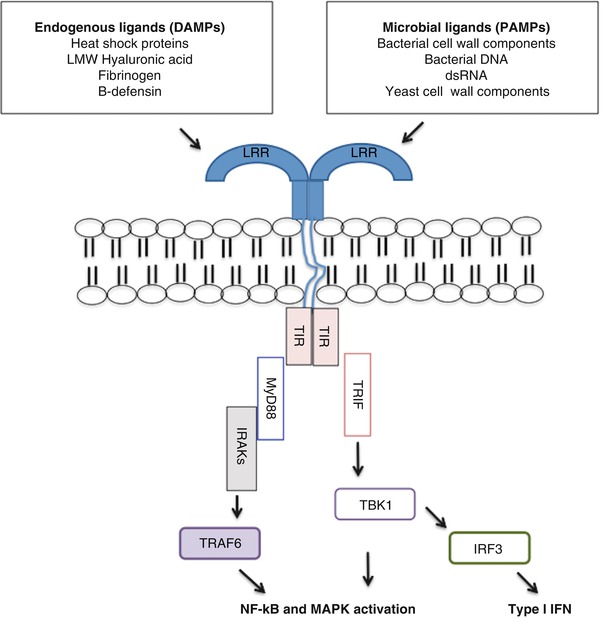
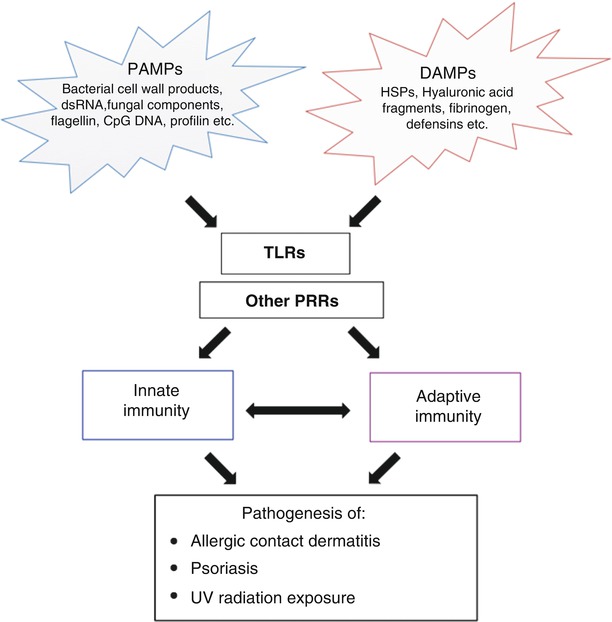
Fig. 2.1
Schematic diagram of TLR activation by various established endogenous and exogenous ligands
Fig. 2.2
The interplay of PAMPs and DAMPs in the activation of TLRs as well as other PRRs. Activation of these receptors influences both arms of immunity and dysregulation of these pathways can lead to inflammation and the development of a variety of dermatological diseases [98]
Table 2.3
TLRs: exogenous ligands (PAMPs) vs. endogenous ligands (DAMPs)
TLR | Exogenous ligands | Endogenous ligands | Signaling pathway | |
|---|---|---|---|---|
1 | Triacyl lipoproteins (w/TLR2) | hBD3 | Heterodimerizes with TLR2; MyD88-dependent signaling | |
2 | Triacyl lipoproteins (w/TLR1) Diacyl lipoproteins lipoteichoic acid, zymosan (w/TLR6) | HMGB1, HSPs, Hyaluronan, Biglycan, Versican, Antiphospholipid antibodies | Heterodimerizes with TLR1 or TLR6; MyD88-dependent signaling | |
3 | dsRNA | Endogenous mRNA from tissue necrosis | TRIP dependent signaling to induce antiviral genes | |
4 | LPS, viral envelope proteins | HMGB1, HSPs, Hyaluronan, Biglycan, Heparan sulphate, hBD2, fibronectin, s100 proteins Fibronectin extra domain A | MyD88 and TRIF/TRAM dependent signaling | |
5 | Flagellin | None identified | MyD88-dependent signaling | |
6 | Diacyl lipoproteins Zymosan Lipoteichoic acids | HMGB1, HSPs, ECM (with TLR2) | Heterodimerizes with TLR2; MyD88-dependent signaling | |
7 | ssRNA | Antiphospholipid antibodies ssRNA | MyD88-dependent signaling | |
8 | ssRNA | Antiphospholipid antibodies ssRNA | MyD88-dependent signaling | |
9 | CpG-DNA | DNA released from acetaminophen-induced hepatoxicity Mitochondrial DNA Immune complexes | MyD88-dependent signaling | |
10 | Unknown | Unknown | MyD88-dependent signaling | [119] |
Toll-Like Receptors in Innate and Adaptive Immunity
As mentioned previously, the pattern recognition theory and identification of TLRs provided the missing link between innate and adaptive immune responses. It is now established that specific ligands activate distinct TLRs and other PRRs, which result in the expression of molecules that shape and fine-tune the adaptive immune response depending on the stimulus involved. On the innate immunity side, activation of TLRs leads to the release of antimicrobial peptides and chemokines that recruit phagocytic cells to the site of infection [120]. TLR activation also induces maturation of dendritic cells to potent APCs via the upregulation of surface expression of MHCII and costimulation markers such as CD80 and CD86 [121].
TLR-mediated effects on the adaptive immune response can be shaped via APCs or T cells directly. It is well known that physical interaction between APCs and T cells requires two signals with signal 1 being the antigen specific signal via MHCII and signal 2 being the expression of costimulation molecules on dendritic cells [122]. TLR stimulation in dendritic cells results in increased expression of MHCII, CD80 and CD86 and is instrumental in promoting both signals required for robust antigen-specific T cell responses [5, 76]. TLR activation on dendritic cells also influences cytokine production, which provides key signals for helper T cell differentiation into different phenotypes with distinct effector functions [123]. For example, TLR-activated dendritic cells produce IFNγ in response to E.coli LPS stimulation which is associated with T helper cell 1 (Th1) differentiation while P. gingivalis LPS induces expression of IL-5, IL-13 and IL-10, cytokines classically associated with Th2 differentiation [124]. Stimulation of APCs with TLR ligands also leads to interleukin-6 (IL-6) secretion, which can result in the loss of suppressor activity by regulatory T cells, allowing for a more effective immune response [125]. Alternatively, TLRs are also expressed in T lymphocytes and TLR ligands can modulate T cell function directly [126]. Direct TLR2 stimulation of T lymphocytes in the absence of APCs has been shown to induce proliferation of regulatory T cells [127]. Intrinsic B cell TLR activation mediates B-cell proliferation and antibody production to T-dependent antigens and similar results were seen in human B cells [128, 129]. Thus, while TLRs are traditionally associated with the innate immune response, they also play key roles in shaping the adaptive immune response and can directly affect the functions of both T and B lymphocytes.
Expression of Human TLRs in Skin
Based on their cellular localization, TLRs can be broadly classified into two groups [84]. TLRs 1, 2, 4, 5 and 6 are expressed on the cell membrane and recognize predominantly microbial membrane components. TLRs 3, 7, 8 and 9, on the other hand, are expressed in intracellular components such as the endoplasmic reticulum, endosomes and lysosomes and primarily recognize microbial nucleic acids. As the primary physical barrier against the environment, it is not surprising that many cell types residing in the skin express a variety of TLRs to survey for pathogens as well as tissue damage signals.
In the epidermis, keratinocytes constitutively express messenger RNA (mRNA) for TLRs 1–6, 9 and 10 [13, 130]. With the exception of TLR10, many studies have demonstrated that keratinocyte TLRs are functional and respond to their respective ligands [130, 131]. Langerhans cells (LCs) express TLRs 1–10 but are most responsive to TLRs 2, 3, 7 and 8 ligands [132, 133]. In the dermis, stimulation of skin/muscle fibroblasts with ligands to TLRs 2, 3, 4, 5 and 9 led to production of specific chemokines [134, 135]. Expression of human TLRs has also been detected on skin resident and trafficking immune cells such as neutrophils, macrophages, dendritic cells, dermal endothelial cells, mucosal epithelial cells, B cells, and T cells (Table 2.4) [133, 145].
Table 2.4
TLR expression in different cell types
Cell type | TLR1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
+ | + | + | + | + | + | + | + | |||
+ | + | + | + | + | + | + | ||||
+ | + | + | + | + | + | + | + | + | + | |
Skin endothelial cells [138] | + | + | + | ++ | + | + | + | + | + | + |
+ | + | + | + | + | ||||||
+ | + | + | + | + | ||||||
MC [141] | + | + | + | + | + | + | + | + | ||
mDCa [142] | + | + | + | + | + | + | + | + | ||
pDC [142] | +/− | +/− | + | + | +/− | |||||
MΦb [143] | + | + | + | + | + | + | + | + | + | + |
N [144] | + | + | + | + | + | + | + | + | + | |
B cell [143] | + | + | + | + | + | + | + | + | ++ | ++ |
+ | + | + | + | + | + | + | + | + | + |
Toll-Like Receptor Signaling
All members of the TLR family are type I transmembrane proteins and contain: (1) extracellular leucine-rich repeats that mediate the recognition of PAMPs, (2) a transmembrane domain and (3) an intracellular tail that contains the Toll/IL-1R (TIR) domain, which bears homology to the IL-1 receptor [84, 146]. Activating ligands lead to homo- or heterodimerization of one TLR with another TLR and result in the dimerization of TIR domains, which serve as the scaffold for downstream adaptor proteins. Important adaptor proteins in TLR signaling include myeloid differentiation factor 88 (MyD88), TIR domain-containing adaptor protein inducing interferon-beta (TRIF) and TRIF-related adaptor molecule (TRAM). MyD88 and TRIF represent distinct signaling pathways that TLRs utilize that result in activation of specific gene programs in response to different activating stimuli.
MyD88 is an adaptor protein that is used by most TLRs with the exception of TLR3 for the initiation of downstream signaling. It should be noted that TLR4 is unique in that its activation results in both MyD88-dependent and TRIF-dependent pathways. In the MyD88 dependent pathway, MyD88 activation results in the recruitment of interleukin-1 receptor-associated kinases 1 (IRAK1) and IRAK4 [147]. IRAK4 then activates IRAK1, leading to IRAK1 autophosphorylation and the dissociation of both members from MyD88 and downstream interaction with tumor necrosis factor receptor–associated factor 6 (TRAF6), an E3 ubiquitin ligase [146]. This signaling complex results in the activation of NF-κB and mitogen-activated protein kinases (MAPKs) and the production of inflammatory cytokines (Fig. 2.1) [84]. Although all TLRs utilize MyD88 as an adaptor protein, it is important to recognize that each TLR utilizes different combinations of adaptor proteins and kinases to generate an immune response that is appropriate for the initial activating stimuli. For instance, activation of TLR2 by lipoproteins leads to TNFα expression while CpG stimulation of TLR9 results in the expression of IFN-α and TNFα [148].
The TRIF-dependent signaling pathway is mainly utilized by TLR3 and TLR4. TLR3 activation results in TRIF recruitment and subsequent activation of TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), a transcription factor required for induction of type I IFNs [148]. TLR4 requires an additional adaptor protein TRAM to stabilize its interaction with TRIF. The discovery of TRIF provided the first molecular explanation for why only TLR3 and TLR4, but not TLR2, can induce IFN-β secretion. Indeed, TRIF-deficient mice were incapable of secreting IFNβ upon stimulation by TLR3 and TLR4 ligands [149]. The TRIF-dependent pathway also results in the activation of NF- κB and MAPKs.
Negative Regulators of TLR Signaling
TLR-mediated signaling plays a key role in the regulation of immunity and excessive TLR signaling has detrimental effects that contribute to autoimmune and inflammatory disease development [150, 151]. Not surprisingly, TLR signaling pathways are tightly controlled and multiple negative regulators of TLR signaling exist at various levels to ensure that immune homeostasis is maintained [100]. IRAK-M, Toll-interacting protein (Tollip) and Suppressor of Cytokine Signaling 1 (SOCS-1) are examples of well-described inhibitors of the TLR signaling pathway. IRAK-M, for instance, is thought to prevent the dissociation of IRAK4 and IRAK1 from MyD88 [152, 153]. Accordingly, IRAK-M−/− macrophages secrete higher levels of inflammatory cytokines and IRAK-M−/− animals are more vulnerable to inflammatory-mediated damage in lupus and lung infection models [154–156]. It is thought that specific genotypes of IRAK-M are associated with sepsis risks (see Table 2.5).
Table 2.5
Negative regulators of Toll-like receptors
Negative regulator | Mechanism of action | Role in human diseases | References |
|---|---|---|---|
Protein regulators | |||
IRAK-M | Prevents IRAK1/IRAK4 dissociation Negatively regulates alternative NF-κB activation after TLR2 stimulation | G/G genotype associated with increased sepsis risk A/A genotype is protective against sepsis Possible role in IBD | |
MyD88s | MyD88 antagonist | Upregulated in septic patients | |
TOLLIP | Autophosphorylates IRAK1 | Polymorphisms mapped in Atopic Dermatitis IBD | |
A20 | De-ubiquitylates TRAF6 | Polymorphisms and mutations associated with rheumatoid arthritis, psoriasis, Sjogren’s Syndrome, SLE, lymphomas | |
SOCS1 | Suppresses IRAK by promoting their degradation | Decreased SOCS1 expression in SLE MS, RA | |
SIGIRR | Orphan receptor that suppresses inflammation | No clear demonstrated role in human disease | |
ABIN-1 | Ubiquitin binding protein that inhibits TLR/C/EBPβ signaling | Protects again psoriasis | |
MicroRNAs Targets 3′-untranslated regions to modulate gene expression | |||
miR-146 | Inhibits IRAK1 and TRAF6 | RA Psoriatic arthritis | |
miR-9 | Blocks NF-κB | Leukemias Cancer | |
miR-21 | Blocks NF-κB and PCDC4 | Cancer | |
miR-155 | Stimulates TNFα Blocks TAK1 activation | Cancer | [180] |
Another negative regulator in the TLR pathway is Toll-interacting protein (TOLLIP), which limits MyD88-dependent NF-κB activation at two different levels [181, 182]. First, overexpression of TOLLIP has been shown to inhibit TLR4– and TLR2–mediated NF-κB activation. TOLLIP also binds directly to IRAK1 to inhibit IRAK1 autophosphorylation and downstream recruitment of signaling proteins required for NF-κB activation [182, 183]. In contrast to IRAK-M−/− mice, TOLLIP deficient animals did not exhibit any overt inabilities to limit the inflammatory response [184]. However, TOLLIP−/− macrophages secreted lower levels of IL-6 and TNFα when stimulated with low doses of LPS, suggesting that TOLLIP is involved in fine-tuning inflammation in response to different levels of stimulation. Polymorphisms of TOLLIP have been associated with atopic dermatitis and inflammatory bowel diseases (see Table 2.5 for other negative TLRs and their association with human diseases). As the role of negative regulators in disease pathogenesis becomes increasingly clear, there is promise that specific targeting of these molecules may lead to the development of new therapeutics.
TLR and Dermatologic Diseases
Acne Vulgaris
Acne vulgaris, a common disorder involving the pilosebaceous unit, is one of the most prevalent conditions in dermatology (see also Chap. 24). It affects more than 45 million people in the United States and is characterized by the presence of inflammatory papules, pustules, nodules and noninflammatory comedones [76, 185]. The pathogenesis of acne is multifactorial but it is generally thought to involve increased sebum production, altered follicular keratinization and an inflammatory response to Propionibacterium acnes, a Gram-positive anaerobe that is a part of normal skin flora, a finding that has been confirmed by recent skin microbiome mapping projects [186, 187]. It is thought that the host immune response [188], and not P. acnes overgrowth, is the main determinant of disease as PBMCs from acne vulgaris patients produce higher levels of IFNγ, IL-12 and IL-8. However, the notion that the host immune response is the main contributor of disease has been challenged by a recent study that showed that acne vulgaris patients harbor different P. acnes strains compared to healthy controls [189].
Early studies demonstrated that soluble factors produced by P. acnes stimulated proinflammatory cytokine production but the exact mechanisms were poorly understood [190, 191]. After the discovery of TLRs, Kim et al. demonstrated thatP. acnes-mediated induction of proinflammatory cytokines was dependent on TLR2 expression and that TLR2 was abundantly expressed on perifollicular macrophages [192]. It was thought that P. acnes possessed two potential cell wall components, LPS and peptidoglycan (PG), that can serve as ligands and activate TLR2 and TLR4 to mediate its downstream proinflammatory response [76]. Indeed, distinct strains of P. acnes with presumably varied modifications in their cell wall components differentially induced upregulation of hBD2, and IL-8 mRNA levels in keratinocytes in a TLR2- and TLR4-dependent manner [45]. Subsequent studies have also found that expression of TLR2 and TLR4 in keratinocytes increased in the epidermis of inflammatory acne lesions and P. acnes exposure led to an increase in TLR2 expression [192, 193]. Other than proinflammatory cytokine production, PAMP stimulation also caused hypercornification of sebaceous glands in a TLR2-dependent manner [17]. While the host immune response is an essential component of acne vulgaris pathogenesis, the molecular mechanisms that differentiate healthy controls and acne vulgaris patients remain poorly characterized. As mentioned earlier, recent studies have showed that different P. acnes strains are found in acne vulgaris patients and there is evidence that these strains can modulate cutaneous innate immunity differentially [189, 194]. Specifically, Jasson et al. demonstrated that only some strains have the capacity to recruit TLR2 receptors and trigger a downstream inflammatory response [194]. It will be interesting to see if the differential capacity of TLR2 recruitment by various P. acnes strains affects keratinocyte proliferation in pilosebaceous units and have clinical implications in acne vulgaris treatment strategies in the future.
Interestingly, retinoids, one of the treatments commonly used for acne vulgaris, have been shown to exert anti-inflammatory effects by decreasing local expression of TLR2 in vitro [18, 19]. These results were recently confirmed in human patients – systemic administration of isotretinoin in acne patients resulted in downregulation of TLR2 cell surface expression on monocytes and decreased levels of IL-1β, IL-6, IL-12 as well as IL-10 release [20]. Of note, systemic isotretinoin decreased TLR2 cell surface expression to levels comparable to those seen in healthy controls. A similar reduction in proinflammatory cytokines was also evident and this effect was sustained for 6 months after the cessation of therapy.
Atopic Dermatitis
Atopic dermatitis (AD) is a common chronic inflammatory skin condition that affects up to 3 % of adults and 15–25 % of children in the United States (see also Chap. 22) [195, 196]. Multiple defects have been identified in AD patients, including impaired skin barrier function, reduced expression of antimicrobial peptides, concomitant skin infections and Th2 skewing. Moreover, it has been demonstrated that up to 90 % of AD patients are colonized with Staphyloccus aureus in both lesional and nonlesional skin, whereas only 5 % of healthy controls exhibit colonization [197]. The molecular details underlying AD pathogenesis are currently under investigation but defects in the TLR signaling pathway have been identified in AD patients. AD patients have decreased TLR2 expression on their circulating monocytes and are impaired in their proinflammatory response to known TLR2 ligands [198, 199]. Werfel and colleagues further reported that a missense mutation in the TLR2 gene (R753Q) is associated with AD patients with a more severe phenotype, higher serum levels of immunoglobulin E (IgE), and greater susceptibility to S. aureus colonization [21–23]. TLR9 and TOLLIP polymorphisms have also been shown to be associated with AD patients [67, 164].
TLRs also directly affect skin barrier function by modulating both physical and chemical properties of barrier function [195]. TLR2 signaling has been shown to increase the expression of tight junction proteins and enhance skin barrier repair [24, 25]. Accordingly, TLR2−/− mice demonstrated impaired repair responses to epidermal injury by tape-stripping, suggesting that TLR2 may contribute to a chronic itch-scratch cycle often seen in AD patients. Other than TLR2, TLR3 signaling in response to dsRNA stimulation from epidermal injury also stimulates the expression of genes involved in permeability barrier repair [26]. In addition, TLR signaling is necessary for the keratinocyte production of antimicrobial peptides (AMPs), a key component of cutaneous chemical barrier function. Previous studies demonstrated that human β-defensin-2 (hBD2) and cathelicidin LL-37 (two AMPs important in keratinocyte defense against S. aureus) were significantly decreased in acute and chronic lesions of AD when compared to controls and patients with psoriasis [200]. LL-37 and hBD2 production, in turn, is dependent on intact TLR2 signaling after S. aureus, S. epidermis and skin injury [201–203].
Consistent with their tendency towards a Th2 immune response, AD patients often suffer from other atopic diseases such as allergic rhinitis, asthma and seasonal allergies. Early lesions in AD have a Th2 cytokine profile, which has been shown in murine models to promote preferential binding to S. aureus [27]. In support of the key role Th2 cytokines (IL-4, IL-13 and TSLP) play in AD pathogenesis, patients with moderate to severe AD treated with dupilumab, an antibody that targets the Th2 cytokine IL-4, showed remarkable improvement in their symptoms [28]. Increasing evidence suggests that TLRs affect the balance between Th1 and Th2 cytokines in the skin. For example, TLR2 stimulation by purified S. aureus-derived diacylated lipopeptitde induces expression of Th2 cytokines like thymic stromal lymphopoietin (TSLP) by keratinocytes [29]. TLR2 ligands also play a role in exaggerating and prolonging Th2-mediated inflammation in AD [26]. TLR2 also has complex roles in modulating other arms of immunity and has been shown to affect mast cell degranulation as well as subsequent IgE antibody production by B cells [30]. Collectively, these data indicate that TLRs, especially TLR2, influence multiple aspects of AD pathogenesis, including barrier function, S. aureus colonization as well as skewing of the immune response towards a Th2 phenotype. Further dissection of how TLRs affect the various altered skin functions in AD will likely lead to development of new therapeutic strategies.
Allergic Contact Dermatitis
Allergic contact dermatitis (ACD) is a common skin disorder caused by type IV delayed hypersensitivity reactions to skin-exposed chemical allergens (see also Chap. 23) [204]. In the clinically silent phase of sensitization, dendritic cells migrate to skin-draining lymph nodes and present contact allergens to naïve T lymphocytes, which may take weeks to months of repeated exposures to low molecular weight compounds. Upon re-exposure to the contact allergen, effector T cells are recruited back to the skin to mediate the type IV delayed hypersensitivity reaction (known as the ‘elicitation phase’) seen in ACD. It is estimated that more than 3000 contact allergens have been described; some of the common contact allergens include nickel, fragrances and hair dyes [98]. Martin et al. [205] first demonstrated a role for TLRs in ACD by showing that mice lacking both TLR2 and TLR4 failed to develop contact hypersensitivity (CHS), the experimental model used to study ACD. Importantly, CHS development was dependent on IL-12 expression that was stimulated by either TLR2 or TLR4 activation of dendritic cells as dendritic cells from TLR2−/− TLR4−/− double knockout animals were resistant to CHS stimulation in wild type animals. Interestingly, CHS developed normally in germ free animals, suggesting that TLR2 and TLR4 activating signals were most likely derived from endogenous ligands such as DAMPs rather than microbial ligands. Further analyses revealed that contact allergens lead to reactive oxygen species (ROS) production, which stimulates the degradation of high molecular weight hyaluronic acid (HA) to low molecular weight HA products [206]. Low molecular weight HA, in turn, can serve as endogenous ligands for TLR2 and 4 signaling and potentiate an inflammatory cascade [207, 208]. A recent study by Gallo and colleagues [209], however, has challenged this notion that HA alone can cause ACD. The group overexpressed hyaluronidase, an enzyme involved in the generation of low molecular weight HA in mice, and showed that small HA fragments alone did not lead to spontaneous cutaneous inflammation resembling CHS. However, the addition of antigen along with small HA fragments accelerated allergic sensitization in a TLR4-dependent manner. Thus, rather than acting as the inflammatory stimuli for ACD, low molecular weight HA controls the antigen presentation capacity of the skin.
Other than DAMP-mediated activation of TLRs, nickel, cobalt and palladium have all been shown to bind and activate human TLR4 [46–48]. Specifically, binding of human TLR4 to nickel was mediated by histidine residues missing in murine TLR4 and provided molecular evidence for why mice are naturally resistant to nickel-induced CHS [48]. Whether nickel alone is sufficient in driving CHS remains unknown although the natural resistance to nicked-induced CHS seen in mice can be overcome by the addition of LPS [210], suggesting that microbial ligands that activate TLR4 may help to amplify the stimulus to promote sensitization to contact allergens [98]. Together, these studies provide evidence that contact allergens like nickel, DAMPS such as low molecular weight HA and PAMPs are all capable of activating TLRs in ACD. However, the relative contribution of each in either the sensitization phase or elicitation phase remains unknown and whether different TLR-expressing skin cells maybe involved in specific phases present exciting future research opportunities for learning more about ACD pathogenesis.
Psoriasis
Psoriasis is a chronic, recurrent, inflammatory disease characterized by dry, scaly, circumscribed erythematous plaques predominantly located in the scalp, nails, extensor surfaces of the limbs, umbilical region, and sacrum (see also Chap. 21). The pathogenesis of psoriasis, which is characterized by the predominance of Th1/Th17 cytokine profiles, involves hyperproliferation and parakeratosis of keratinocytes, which ultimately leads to thickening of the epidermis [99]. Many advances have been made in understanding the mechanisms involved in psoriasis and developments of new immunosuppressive and biologic treatments. Not surprisingly, TLRs have also been found to play a role in the pathogenesis of psoriasis. A study demonstrated that TLR1 and TLR2 expression was increased in the suprabasal layer of keratinocytes in psoriasis patients compared to skin isolated from normal controls [13]. In contrast, TLR5 expression in basal keratinocytes from psoriatic patients was decreased compared to healthy controls. Other studies have found increased TLR1, 2, 4, 5 and 9 expression in keratinocytes isolated from psoriatic lesions [211]. A recent study also identified mutations in the gene AP1S3, a protein involved in TLR3 trafficking, that are associated with pustular psoriasis [43]. Furthermore, application of imiquimod, a known TLR7 agonist, is known to trigger psoriasis in both humans and animal models [55, 56]. It is thought that imiquimod activates TLR7 signaling on DCs to drive psoriatic plaque formation by activating the production of IL-17 and IL-22 by innate lymphoctyes. ABIN-1, a negative regulator of TLR signaling, protects against psoriasis development by preventing exaggerated NF-κB and MAPK signaling in response to TLR7 agonists [172]. Therefore, TLR expression on various cell types in the skin may drive psoriatic pathogenesis and it is plausible that different cell types maybe involved in different phases of disease progression.
In contrast to AD patients who are more susceptible to S. aureus infections (see above), it is generally accepted that psoriatic plaques are relatively resistant to S. aureus infection [212]. It is thought that increased AMP production such as hBD2 and syndecans seen in psoriatic plaques is partially responsible for this phenotype [213, 214]. Keratinocyte growth factor, TGFα, has been found at high levels in psoriatic lesions and is responsible for increased TLR5 and TLR9 expression as well as TLR-dependent release of AMPs and proinflammatory cytokines [215]. While the increased production of AMPs is beneficial against pathogenic microorganisms, it has been postulated that they may also contribute to inflammation by modulating host immune receptors such as TLRs [185]. For example, LL-37 has been shown to complex with self DNA to create a novel DAMP and activate plasmacytoid dendritic cells (pDCs) via the TLR9 pathway and drive inflammation in psoriatic skin by stimulating IFNα production [68]. A recent study showed that LL-37 and an alternatively processed cathelicidin peptide KS-30 also stimulate keratinocytes to produce more type I IFNs but this was not dependent on its complexed DNA that was important for pDC activation [216].
Other than AMPs, heat shock protein (HSP) expression is also thought to contribute to TLR-mediated inflammation. HSP is induced by exposure to microbial pathogens and other stressful stimuli [49]. Heat shock protein 27, 60, 70 and 90 have been shown to be overexpressed in psoriasis [49, 50] and can trigger an innate immune response through TLR4 on APCs, resulting in the secretion of TNFα, IL-12, and other Th1 cytokines. They also may act on the adaptive immune response by serving as autoantigens for self-reactive T cells that migrate into psoriatic lesions.
These discoveries are opening doors for novel treatments in psoriasis (see Chaps. 43). It is thought that systemic and topical retinoids used in the treatment of psoriasis may control inflammation through their inhibitory effects via TLR2 [76]. Monomethylfumarate (MMF), a bioactive metabolite of fumaric acid ester, is an immunotherapy for psoriasis that causes decreased production of Th1 cytokines and lymphocytopenia [217]. Monomethylfumarate was shown to decrease DC response to LPS and decreased IL-12p70 and IL-10 production. Etanercept, a TNFα inhibitor that has been successful in psoriasis treatment, has been shown to be associated with decreased LL-37 expression, which may dampen TLR9 activation and further suppress the chronic inflammatory response in psoriasis [218]. Thus, TLR dysregulation appears to play a role in psoriasis pathogenesis although whether a predominant TLR is involved remains unclear. Continued research in these areas will yield interesting findings that will impact treatment options for psoriasis patients.
Bacterial Infections
Bacterial cell wall components were the original ligands shown to stimulate TLR signaling [80, 81]. Accordingly, TLRs have been implicated in the pathogenesis of multiple bacterial diseases.
S. aureus Infections
S. aureus, a gram-positive extracellular bacteria, is the causative agent of a variety of skin infections, including impetigo, folliculitis and cellulitis (see Chap. 16) [219]. It is estimated that 20 % of the population is persistently colonized, harboring S. aureus on the skin and the nares, while 50 % are intermittent carriers [185]. S. aureus lipoproteins, peptidoglycan and lipoteichoic acid signal through TLR2/6 and TLR2/2 dimers [220, 221]. Accordingly, TLR2 deficient mice were more susceptible to S. aureus infection and harbored higher bacterial loads in blood compared to wild type controls [31, 32]. Animals deficient in MyD88, the key adaptor protein required for all TLR signaling with the exception of TLR3, were also more susceptible to S. aureus infection and demonstrated a neutrophil recruitment defect that was not seen in TLR2−/− mice. In corroboration of these animal studies, MyD88-deficient and IRAK4-deficient patients are more susceptible to S. aureus infections [222]. Mutations in the IRAK4 kinase that led to premature stop codons have been shown to increase susceptibility to pyogenic infections caused by S. aureus as well as Streptococcus pneumonia [223]. Cells from patients with this disease did not respond to any known ligands from TLRs 1 to 6 and 9. Consistent with an immune deficient phenotype, these patients suffered recurrent pyogenic infections with minimal febrile or inflammatory responses.
Leprosy
Leprosy, or Hansen’s disease, caused by Mycobacterium leprae, is a chronic, debilitating disease that encompasses a spectrum of clinical manifestations [76]. At one end, tuberculoid leprosy (TL) presents in patients with a strong cell-mediated immune response, resulting in high resistance to M. leprae and few, localized, paucibacillary lesions. At the other end of the spectrum, lepromatous leprosy (LL) patients have a weak immune response, resulting in disseminated, multibacillary disease, including cutaneous and nerve involvement [224]. Other forms of the disease with unstable resistance include borderline tuberculoid, borderline, and borderline lepromatous. The former is Th1 mediated (e.g., IFNγ, IL-12, IL-18, and granulocyte-macrophage colony-stimulating factor), whereas the latter is Th2 driven (e.g., IL-4 and IL-10). There is accumulating evidence to suggest that whether a patient develops one response over the other may be in part due to variations in the TLR signaling pathway.
In 1999, it was discovered that mycobacteria activated macrophages through TLR2, resulting in production of TNFα, a proinflammatory cytokine [225]. An introduction of a dominant negative mutation in TLR2 rendered the receptor unresponsive to M. tuberculosis. Furthermore, a mutation in Arg677Trp in TLR2 has been associated with LL in the Korean population [33]. A separate study confirmed that this mutation halts the ability of TLR2 to respond to both M. leprae and M. tuberculosis, confirming the clinical importance of this polymorphism [224].
Upon stimulation with M. leprae, patients with the Arg677Trp TLR2 mutation were found to have decreased production of IL-2, IL-12, IFNγ, and TNFα, and increased IL-10 (an anti-inflammatory cytokine) when compared to those with the wild-type TLR2 [34]. Thus, the mutated TLR2 favored a Th2 phenotype, which is consistent with the observed LL phenotype. Based on these findings, TLR2 appears to play a critical role in the alteration of cytokine profiles and determination of the type of leprosy that develops.
M. leprae products were shown to activate both TLR2 homodimers as well as TLR1-TLR2 heterodimers [11]. Interestingly, TL lesions had higher TLR1 and TLR2 expression compared to LL lesions, suggesting that the expression of TLR2 and TLR1 contributes to the host response. Moreover, this study demonstrated that type 1 cytokines enhance TLR1 and TLR2 activation, whereas the Th2 cytokines inhibited activation. Therefore, not only does innate TLR signaling affect the adaptive immune response, but also the adaptive immune response, through cytokine release, may also influence the innate response. Further evidence that TLRs play a role in M. leprae pathogenesis was shown in a recent genetic study. Wong et al. showed that individuals homozygous for the TLR1 I602S mutation, a functional TLR1 knockout, were protected from M. leprae infection, suggesting that M. leprae may have utilized TLR1 signaling to enhance its pathogenesis [12]. These findings underline the complexity of the interaction between TLRs and M. leprae pathogenesis through evolution and provide additional proof that TLRs are involved in bridging the gap between innate and adaptive immunity.
Syphilis
Syphilis is a contagious, sexually transmitted disease caused by the obligate human pathogen Treponema pallidum [76]. There are three stages of syphilis. In primary syphilis, a painless genital ulcer, called a chancre, appears 18–21 days after infection. Secondary syphilis can appear as various cutaneous eruptions—macular, papular, or polymorphous—often with lesions on the palms and soles. Tertiary syphilis occurs 3–5 years after infection. Patients may develop gummas, or necrotic lesions in the skin, mucous membranes, bones, or joints. Other complications of syphilis include neurologic and cardiac involvement.
It is appreciated that the outer cell wall structures of spirochete bacteria like T. pallidum are vastly different from the typical outer membranes of Gram-negative bacteria [226]. It is thought that T. pallidum has developed multiple strategies to evade the host immune response. For instance, T. pallidum lacks LPS and contains a paucity of immunogenic proteins compared to other spirochete bacterium [227]. Thus, during syphilitic infection, T. pallidum membrane lipoproteins (LPs) serve as principal proinflammatory mediators [35]. Indeed, it was demonstrated that T. pallidum LPs stimulated TLR2- and TLR4-expressing immature murine dendritic cells (DCs) to release proinflammatory cytokines such as IL-12, IL-1β, TNFα, and IL-6. It was long thought that opsonization of spirochete bacteria was essential for T. pallidum clearance but mechanistic studies were missing until Silver et al. recently demonstrated that TLR-MyD88 signaling is crucial for phagocytosis and bacterial clearance [227]. MyD88-deficient animals exhibited increased inflammation with a stronger infiltration of neutrophils and lymphocytes but still harbored a high bacterial load due to the inability of MyD88−/− macrophages to opsonize T. pallidum. Consistent with these findings, a recent clinical study found that TLR1, TLR2 and TLR6 polymorphisms are associated with an increased risk of neurosyphilis development, suggesting that the TLR1/TLR2 and TLR2/TLR6 heterodimers are important in protecting against T. pallidum [16].
Yersinia pestis
Y. pestis is a gram-negative bacillus that causes plague, a disease that killed millions of people in the “Black Death” pandemic. It is transmitted by the bite of the rat flea Xenopsylla cheopis. Clinically, painful buboes form in the axillae or groin, although other skin lesions such as vesicles, plaques, petechiae, and purpura can be seen. Yersinia outer membrane protein, V antigen, targets TLR2 and CD14 on the surfaces of APCs [228]. Interestingly, Y. pestis has specific variations in its LPS lipid A structure to evade TLR4-mediated host immune recognition [229].
Lyme Disease
Lyme disease is a tick-borne illness caused by the spirochete Borrelia burgdorferi and is loosely divided into three stages. The primary stage is characterized by constitutional symptoms and erythema chronicum migrans. The second stage occurs for 5–6 months after the rash resolves. In the tertiary phase, cardiac, neurologic, and rheumatologic complications can occur. Like other spirochetes such as T. pallidum, B. burgdorferi does not have LPS in its outer membrane structure to stimulate TLR4. B. burgdorferi outer surface protein A (OspA) stimulates TLR2 to activate inflammatory signaling [36]. Stimulation with B. burgdorferi lysate was found to increase the expression of TLR1 and TLR2 in all peripheral blood monocytes and human brain cells, but not neurons [230]. Consistent with the aforementioned in vitro data, TLR2 deficient animals harbored much higher loads of B. burgdorferi and TLR2−/− macrophages produced lower levels of proinflammatory cytokines [231]. Peripheral blood monocytes (PBMCs) isolated from patients with TLR2 Arg753Gln mutations also secreted less proinflammatory cytokines [37]. Interestingly, the lower levels of TNFα and IFNγ were protective against late stages of disease such as lyme arthritis development.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


