Fig. 35.1
Diagram of the hemidesmosome. Individual proteins comprise the hemidesmosome and many of these proteins serve as antigenic targets in clinical variants along the pemphigoid spectrum
Characterization of autoantibodies reveals a polyclonal response, with the majority of antibodies recognizing a cluster of epitopes about the largest noncollagen domain (referred to as NC16A) of the BP180 antigen [37, 38]. The BP180NC16A specific autoantibodies are predominantly IgE, IgG1, and IgG4 [39–41]. Titers of IgG and IgE BP180NC16A autoantibodies correlate to the degree of clinical severity [40–42]. Furthermore, autoantibodies against the BP180NC16A domain are sufficient to induce subepidermal blisters in the neonatal passive transfer model [43].
B cell production of pathogenic autoantibodies likely begins with development of autoreactive T cells. Most patients with BP demonstrate circulating autoreactive BP180-specific CD4 T cells as well as T-helper 2 (Th2) and Th1 responses against the BP180 ectodomain [44, 45]. Furthermore, BP180-reactive Th cells and IgG autoantibodies recognize similar or identical epitopes clustered in distinct regions of the BP180 ectodomain; the majority of autoreactive Th2 and Th1 cells and B cells recognize epitopes within the NC16A, followed by reactivity against the COOH-terminal and central regions [45–48]. Interestingly, T and B cell reactivity against the BP180 NC16A ectodomain is associated with severe BP, with widespread blisters and erosions, while responses against the central portion is more common in limited BP, with few blisters and erosions [47]. Less than 50 % of BP patients also show a combined T and B cell response against the COOH- and NH2-terminal globular domains of BP230 [48].
The characteristic histologic and clinical features of BP are a result of the inflammatory cascade triggered by the binding of pathogenic autoantibodies to the antigen targets within the hemidesmosome structure. In vitro and in vivo studies show that antibodies against BP180 are pathogenic [7, 49]. In the in vivo model, neonatal mice injected intraperitoneally or intradermally with anti-murine BP180 antibodies develop BP-like skin lesions, including in situ deposition of IgG and complement C3 at the BMZ and an inflammatory cell infiltrate. Subepidermal blistering in this IgG passive transfer model of BP requires complement activation, mast cells and neutrophils [50–53]. Inflammatory cell products, such as neutrophil elastase, gelatinase and plasmin, result in tissue destruction at the dermal-epidermal junction, which manifests histologically as a sub-epidermal split [54–58].
Although the IgG autoantibodies in BP have been most studied to date, there is increasing evidence that IgE autoantibodies may also play a role in the pathogenesis of BP. As previously mentioned, over 90 % of BP patients have detectable circulating IgE anti-BP180 autoantibodies, and titers of IgE anti-BP180NC16A autoantibodies correlate with disease activity [40, 59]. IgE anti-BP180 autoantibodies likely play a role in the pathogenesis of BP by activation of mast cells and recruitment of eosinophils, as shown in murine models [60, 61]. Additional evidence comes from the dramatic improvement of several recalcitrant BP patients treated with the omalizumab, a monoclonal antibody that inhibits IgE binding to the high affinity IgE receptor [62, 63].
Clinical Features
Clinical manifestations of BP include pruritic, large, tense bullae overlying urticarial plaques (Fig. 35.2) [1]. However, over half of BP patients present with nonbullous lesions, as the prodromal phase is typically non-specific [64]. Intense pruritus, with or without an eczematous or urticarial eruption, are typically the first signs of the disease. In most patients, the prodromal phase is followed by the development of symmetric tense bullae, distributed primarily on the flexural aspects of the extremities and lower trunk. The bullae are symptomatically pruritic, and may be filled with clear or hemorrhagic fluid. Rupture of the bullae results in shallow crusted erosions. Lesions heal without scarring, though milia formation may occur and postinflammatory pigmentation is quite common. Involvement of intertriginous areas may produce vegetative moist plaques. The mucous membranes are involved in a minority of patients (less than 10 %) and lesions are typically limited to the oral mucosa [1, 65, 66].
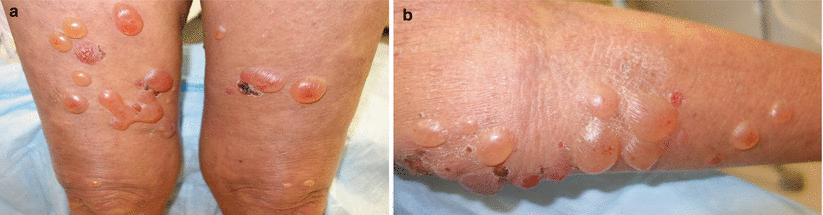
Fig. 35.2
BP classically presents with tense bullae overlying urticarial plaques on the trunk and extremities lower extremities (a) upper extremity (b) of elderly patients
Diagnosis
Once the suspicion of BP is raised on the basis of clinical and historical patient information, definitive diagnosis is based not only on histologic evaluation, but also on further characterization of the autoantibody response through immunological techniques including direct and indirect immunofluorescence (IF) microscopy and enzyme linked immunosorbant assay (ELISA). Standard histologic evaluation via light microscopy reveals a subepidermal split with inflammatory infiltrate composed mainly of eosinophils and neutrophils in the upper dermis [1]. Occasionally, cell poor BP is observed where the inflammatory infiltrate is minimal or absent. Eosinophilic spongiosis has also been described in BP [67].
Direct IF (from perilesional, uninvolved skin) demonstrates a fine linear band at the dermal-epidermal junction of IgG and/or C3 with a sensitivity of 91 % [68] (Fig. 35.3a). Indirect IF (from patient’s serum) demonstrates autoantibodies that bind the dermal-epidermal junction in a linear fashion with a sensitivity of roughly 70 %. The use of salt split skin as the substrate increases the sensitivity of indirect IF and also allows for distinction between BP (with localization of the autoantibodies to the epidermal side of salt split skin) and epidermolysis bullosa acquisita (with localization of autoantibodies to the dermal side of salt split skin) (Fig. 35.3b) [69, 70]. The BP180 NC16A domain ELISA is emerging as a more readily available test to evaluate for circulating autoantibodies in the serum and is being utilized in both the clinical and research setting. ELISA index values tend to correlate with disease activity with a sensitivity ranging from 72 to 89 % [68, 71]. However, approximately 7 % of the normal population has detectable anti-BP180 autoantibodies by ELISA without regard to age [68, 72]. The relevance of a positive BP180 ELISA in the absence of clinical findings is unclear, but the 7 % false positive rate suggests that ELISA should be used in the appropriate clinical context and with supporting direct and indirect IF studies rather than as the sole immunological criteria for diagnosis.
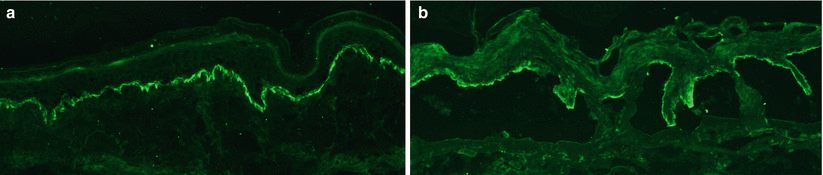
Fig. 35.3
Direct and indirect immunofluorescence (IF) findings of BP. (a) Direct IF shows C3 and IgG deposition in a linear pattern along the basement membrane zone. (b) Indirect IF shows IgG localization to the epidermal side of salt split skin
Therapy
Topical high potency corticosteroids are useful for limited areas of involvement in motivated patients. More generalized or severe manifestations typically require systemic corticosteroids, in initial dosing ranging from 0.5 to 1 mg/kg/day. After gaining control of development of new bullae, the dose may be tapered over 6–8 months, with dosing adjusted to account for the recurrence of disease. A recent study suggests that high potency topical steroids (clobetasol) may be an excellent approach to induce clinical remission in BP [24, 73].
In cases refractory to systemic corticosteroids, additional immunosuppressive medications may be employed. Methotrexate, azathioprine (dose adjusted for variability in thiopurine methyltransferase levels) and mycophenolate mofetil are all options, with selection based upon each medication’s side effect profile [74–79]. Dapsone may be useful in some cases where systemic steroids or immunosuppressive drugs are contraindicated [80–82]. Plasmapheresis and intravenous immunoglobulin have been used successfully to induce remission in BP [83–85]. Rituximab, a monoclonal anti-CD20 antibody that targets B lymphocytes, has also shown efficacy in treating bullous pemphigoid [86]. In a small cohort of patients, low serum B cell activating factor (BAFF) levels and a higher proportion of memory B lymphocytes during B cell recovery post treatment were seen in those patients that flared post treatment [87]. While other dermatologists have found tetracycline family members and nicotinamide useful in the control of BP, in our hands it has had a limited effect in some rare patients.
Cicatricial Pemphigoid
Overview
Cicatricial Pemphigoid (CP), also known as mucous membrane pemphigoid, is a rare chronic autoimmune blistering disorder, characterized by mucosal involvement and a high risk of scarring within the affected areas. Any or all mucous membranes may be involved in any individual patient. Direct IF demonstrates linear deposition of autoantibodies against various basement membrane zone components. The clinical course tends to be chronic and best managed with a multi-disciplinary approach to avoid major sequelae secondary to scarring [88].
Historical Background
Chronic blistering conditions with both skin and eye involvement were described throughout the 1800s [89, 90]. Up to the mid-1900s, CP was considered a variant of pemphigus, until Civatte and Lever separated the disorder based on histopathologic criteria [91, 92]. Lever suggested changing the name from “benign mucous membrane pemphigoid” to “cicatricial pemphigoid” based upon the tendency for scar formation [1, 93]. Tissue-bound immunoglobulins and complement components along the basement membrane zone were identified in the 1970s [94, 95] and circulating antoantibodies were demonstrated by Dantzig and Bean shortly thereafter [96, 97].
Epidemiology
CP has been documented to occur at any age, and is one of the most rare subepidermal bullous diseases, with an estimated annual incidence of approximately 1 per million per year [16, 18, 98]. There is a female predilection with females being affected twice as frequently as males with onset typically in the 60s [16, 18, 99]. In a recent review of patients with circulating antibodies recognizing laminin-5, over 30 % of patients demonstrated internal malignancy, suggesting an increased relative risk of developing malignancy in this type of CP [100–102].
Pathogenesis
While CP is clinically characterized by the unifying clinical feature of mucosal involvement and scar formation and immunofluorescence finding of autoantibody deposition at the basement membrane zone, the antigenic targets can be quite heterogeneous. Commonly recognized target antigens in CP include BP180, BP230, laminin-332 (also known as laminin-5, or epiligrin), laminin-6, integrin ß4 subunit, and integrin α subunit [103–106]. While autoantibodies are typically of the IgG class, IgA autoantibodies are also detected in some patients as well. The collective contribution of how the autoantibodies, the specific antigenic targets, and the microenvironment lead to mucosal-predominant inflammation and scar formation is still not understood.
Clinical Features
While any mucosal surface may be affected in CP, the oral site is most commonly involved, followed by, in decreasing order of involvement, the ocular, nasal, nasopharyngeal, anogenital, laryngeal, and esophageal sites [107–109]. Oral disease may present as erosive or desquamative gingivitis; intact blisters are rarely observed (Fig. 35.4a). Chronic erosions on the palate and lateral tongue are commonly seen, as well. Once healed, the areas may resemble lesions of lichen planus, with white reticulated striations.
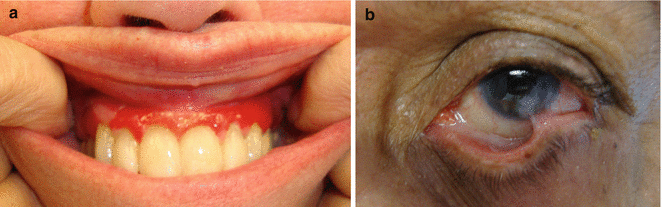
Fig. 35.4
Clinical manifestations of cicatricial pemphigoid (CP). (a) Oral manifestations of CP often include desquamative gingivitis. (b) Ocular manifestations of CP can include symblepharon formation
Conjunctival involvement is also commonly seen, and, if not appropriately addressed, may result in blindness. Conjunctival inflammation, erosions, symblepharon, entropion and trichiasis may be present (Fig. 35.4b). Symptoms include dryness, burning and foreign body sensation of the eyes. Similar to the appearance of lesions in the oral mucosa, intact bullae are very rarely observed [107]. CP may be limited to the ocular mucosa in some patients without other sites of involvement. Examination by an ophthalmologist is critical for detection of early disease by slit-lamp evaluation.
Nasopharyngeal involvement may be associated with lesions of the upper aerodigestive tract [108]. Symptoms suggestive of nasopharyngeal involvement include nasal crusting, recurrent epistaxis, and nasal airway obstruction. A history of persistent pharyngalgia, dysphagia, odynophagia, dysphonia, or dyspepsia may suggest pharyngeal and laryngeal involvement [110]. Development of strictures or stenosis of the pharynx or larynx may prove life threatening, if not identified at an early stage.
Genital and anal involvement is relatively rare. Progressive disease may lead to narrowing of the introitus in women, or to phimosis in men. Anal involvement may lead to scarring, and potentially to stricture. The skin is involved in approximately a quarter of patients with CP with lesions most frequently appearing on the head, neck, and upper trunk. Erythematous plaques develop recurrent blisters, and heal with atrophic scarring.
Diagnosis
Diagnosis of CP relies on immuofluorescent evaluation. Direct IF performed on perilesional mucosa or skin reveals IgG, IgA, and/or C3 in a continuous fine linear pattern along the basement membrane zone [88, 111–114]. Direct IF is most likely to provide conclusive information when performed on an area of mucosa adjacent to an area of inflammation [88]. In order to minimize the risk of conjunctival scarring, other anatomic areas of involvement should be biopsied preferentially, as injury to the conjunctiva may increase disease activity [88]. Indirect IF reveals autoantibodies at the basement membrane zone in 20–30 % of patients with clinical disease. Localization of autoantibodies, when performed on human salt-split skin substrate, is variable, depending upon the diversity of each patient’s autoantibody milleu and the location of the autoantigens targeted [70, 115–119].
Therapy
In patients with mild, low-risk disease (defined as oral mucosa involvement, with or without skin involvement), potent topical corticosteroids may be sufficient. However, in patients with high-risk disease (defined as involvement of ocular, genital, laryngeal, nasopharyngeal, esophageal sites), initial therapy should involve systemic corticosteroids, with consideration of addition of cyclophosphamide or azathioprine [88]. Dapsone is also a consideration for stable or mild disease. Case reports document the use of various other modalities, including intravenous immunoglobulin, mycophenolate mofetil, and most recently, rituximab [120–125].
Herpes Gestationis (Pemphigoid Gestationis)
Overview
Herpes Gestationis (HG) is a rare dermatosis of pregnancy and the immediate post-partum period characterized by intensely pruritic urticarial lesions and eventually tense bullae. Most patients demonstrate autoantibodies to the BP180 antigen, with generation of a subepidermal separation on histologic evaluation. In pregnancies affected with HG, there is increased risk of prematurity and small for gestational age birth. The clinical course tends to be quite variable, and most cases resolve following delivery.
Historical Background
The term herpes gestationis was first cited by Milton in 1872, with demonstration of complement deposition along the basement membrane zone first documented by Provost in 1973 [126]. In 1976, Jordon et al and Katz et al characterized the “HG” factor as an IgG antibody that activates the complement pathway [127, 128]. Guidice et al identified the structural antigen for the “HG factor” as BP180, and provided further structural analysis of the antigen [37, 129].
Epidemiology
HG is a rare disorder exclusively seen during late pregnancy and the immediate post-partum period. Even more rare is its association with trophoblastic malignancy or molar pregnancy [130, 131]. Estimates of incidence for HG range from 1 in 10,000 to 1 in 50,000 pregnancies [132, 133]. HG often recurs in subsequent pregnancies, and may occur earlier and in a more severe form [134]. Also, with subsequent pregnancies, the time to resolution in the post-partum period may become progressively prolonged [135].
Pathogenesis
Generation of autoreactive T cells stimulate production of autoantibodies against the NC16A region of the BP180 antigen, prompting histopathologic and clinical manifestations of HG. Characterization of the autoreactive T cells has revealed expression of a Th1 cytokine profile, supporting the production of IgG1 autoantibodies against BP180 [136, 137].
Deposition of the autoantibodies prompts complement activation, primarily through the classical pathway, followed by mast cell recruitment, degranulation, recruitment of inflammatory cells (primarily eosinophils), and production of destructive proteases [138–140]. The resultant damage at the dermal-epidermal junction manifests as a subepidermal separation on histologic examination.
Clinical Features
Typically, abdominal urticarial lesions appear abruptly during the second or third trimester, followed by the appearance of a generalized bullous reaction sparing the face, mucous membranes, palms and soles [133, 134, 145, 146]. However, initial onset in the immediate postpartum period has been described in 20 % of cases [147]. Most patients experience a flare with delivery, which spontaneously resolves over weeks to months following delivery. Flares with subsequent pregnancies, menstruation, or initiation of oral contraceptive use are common [145, 148].
Neonatal vesicles are present in approximately 10 % of cases, presumably secondary to passive transfer of pathogenic antibodies [145, 149, 150]. While the lesions are transient and typically mild, they are at increased risk of superinfection secondary to the infant’s relatively immunocompromised status. Although associations have been reported in regards to preterm delivery and small for gestational age infants, there have been no reports of increased fetal morbidity or mortality [151, 152].
Diagnosis
Standard light microscopy of involved skin reveals a subepidermal vesicle with a lymphocytic and eosinophilic perivascular infiltrate. Direct IF of perilesional skin demonstrates linear C3 along the basement membrane zone; occasionally, IgG is also present, but to a lesser degree than C3 [140, 153]. While standard indirect IF is rarely positive, complement-added indirect IF is almost universally positive for pathogenic IgG antibodies [153].
Therapy
For limited disease, high potency topical corticosteroids along with oral antihistamines may provide sufficient control. However, more severe eruptions may require oral corticosteroids for adequate response. Intravenous immunoglobulin has also been effective for some patients [154]. More therapeutic options are feasible during the postpartum period, such as cyclophosphamide and methotrexate, although only sporadic reports of variable efficacy are available [155, 156].
Summary
The pemphigoid spectrum of diseases shares histological and immunofluorescence features of subepidermal blister formation with detection of autoantibodies and complement along the basement membrane zone. While BP180 is the antigenic target in the most classic form of the disease, clinical variants exist with more heterogeneous target autoantigens. The diagnosis of these diseases relies upon the triad of supporting clinical, histological, and immunological features. Treatment is aimed at suppressing the misguided immune response with topical and/or systemic corticosteroids in addition to other immunosuppressive agents. Progress continues to be made in understanding disease initiation, pathogenesis and more targeted treatment strategies.
Questions and Answers
- 1.
What diseases are associated with bullous pemphigoid?
Patients with neurological diseases including dementia, stroke, and Parkinson’s disease show an increased risk of developing BP
- 2.
Describe the stepwise events involved in the pathogenesis of bullous pemphigoid.
The pathogenesis of bullous pemphigoid starts with autoantibody formation against BP180 and BP230. Bound autoantibodies fix complement at the basement membrane zone and elicit an inflammatory cell infiltrate of neutrophils, mast cells, and eosinophils. Inflammatory cell products, such as neutrophil elastase, gelatinase and plasmin, result in tissue destruction at the dermal-epidermal junction, which manifests histologically as a sub-epidermal split
- 3.
What are the most common sites of involvement in cicatricial pemphigoid?
While any mucosal surface may be affected in CP, the oral site is most commonly involved, followed by, in decreasing order of involvement, the ocular, nasal, nasopharyngeal, anogenital, laryngeal, and esophageal sites. The skin is involved in a minority of patients
References
1.
Lever WF. Pemphigus. Medicine (Baltimore). 1953;32(1):1–123.
2.
Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WL, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA. 1967;200(9):751–6.PubMed
3.
Stanley JR, Hawley-Nelson P, Yuspa SH, Shevach EM, Katz SI. Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell. 1981;24(3):897–903.PubMed
4.
Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol. 1986;136(4):1231–5.PubMed
5.
Mutasim DF, Takahashi Y, Labib RS, Anhalt GJ, Patel HP, Diaz LA. A pool of bullous pemphigoid antigen(s) is intracellular and associated with the basal cell cytoskeleton-hemidesmosome complex. J Invest Dermatol. 1985;84(1):47–53.PubMed
6.
Diaz LA, Ratrie 3rd H, Saunders WS, et al. Isolation of a human epidermal cDNA corresponding to the 180-kD autoantigen recognized by bullous pemphigoid and herpes gestationis sera. Immunolocalization of this protein to the hemidesmosome. J Clin Invest. 1990;86(4):1088–94.PubMedPubMedCentral
7.
Liu Z, Diaz LA, Troy JL, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. 1993;92(5):2480–8.PubMedPubMedCentral
8.
Rzany BWN. Epidemiology of autoimmune skin disorders. In: Michael Hertl. Autoimmune diseases of the skin. New York: Springer; 2001. p. 21–38.
9.
Bastuji-Garin S, Joly P, Lemordant P, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol. 2011;131(3):637–43.PubMed
10.
Langan SM, Groves RW, West J. The relationship between neurological disease and bullous pemphigoid: a population-based case-control study. J Invest Dermatol. 2011;131(3):631–6.PubMed
11.
Cordel N, Chosidow O, Hellot MF, et al. Neurological disorders in patients with bullous pemphigoid. Dermatology. 2007;215(3):187–91.PubMed
12.
Jedlickova H, Hlubinka M, Pavlik T, Semradova V, Budinska E, Vlasin Z. Bullous pemphigoid and internal diseases – a case-control study. Eur J Dermatol. 2010;20(1):96–101.
13.
Stinco G, Codutti R, Scarbolo M, Valent F, Patrone P. A retrospective epidemiological study on the association of bullous pemphigoid and neurological diseases. Acta Derm Venereol. 2005;85(2):136–9.PubMed
14.
Lindelof B, Islam N, Eklund G, Arfors L. Pemphigoid and cancer. Arch Dermatol. 1990;126(1):66–8.PubMed
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


