Introduction36
INTRODUCTION
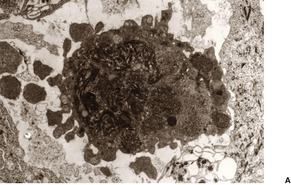
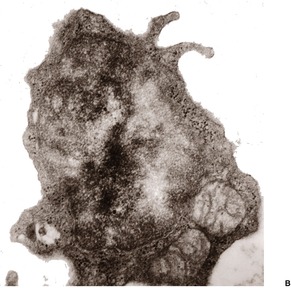
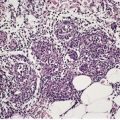
Fig. 3.1
Disease
Histopathological features
Lichen planus
Prominent Civatte bodies, band-like inflammatory infiltrate, wedge-shaped hypergranulosis. Hypertrophic form has changes limited to the tips of the acanthotic downgrowths and often superadded lichen simplex chronicus. The infiltrate extends around hair follicles in lichen planopilaris. Pigment incontinence is conspicuous in erythema dyschromicum perstans.
Lichen nitidus
Focal (papular) lichenoid lesions; some giant cells; dermal infiltrate often ‘clasped’ by acanthotic downgrowths.
Lichen striatus
Clinically linear; irregular and discontinuous lichenoid reaction; infiltrate sometimes around follicles and sweat glands.
Lichen planus-like keratosis
Solitary; prominent Civatte body formation; solar lentigo often at margins.
Lichenoid drug eruptions
Focal parakeratosis; eosinophils, plasma cells and melanin incontinence may be features. Deep extension of the infiltrate occurs in photolichenoid lesions.
Fixed drug eruptions
Interface-obscuring infiltrate, often extends deeper than erythema multiforme; cell death often above basal layer; neutrophils often present.
Erythema multiforme
Interface-obscuring infiltrate; sometimes subepidermal vesiculation and variable epidermal cell death.
Graft-versus-host disease
Basal vacuolation; scattered apoptotic keratinocytes, sometimes with attached lymphocytes (‘satellite cell necrosis’); variable lymphocytic infiltrate.
Lupus erythematosus
Mixed vacuolar change and Civatte bodies. SLE has prominent vacuolar change and minimal cell death. Discoid lupus away from the face has more cell death and superficial and deep infiltrate; mucin; follicular plugging; basement membrane thickening. Some cases resemble erythema multiforme with cell death at all layers.
Dermatomyositis
May resemble acute lupus with vacuolar change, epidermal atrophy, some dermal mucin; infiltrate usually superficial and often sparse.
Poikilodermas
Vacuolar change; telangiectasia; pigment incontinence; late dermal sclerosis.
Pityriasis lichenoides
Acute form combines lymphocytic vasculitis with epidermal cell death; interface-obscuring infiltrate; focal hemorrhage; focal parakeratosis.
Paraneoplastic pemphigus
Erythema multiforme-like changes with suprabasal acantholysis and clefting; subepidermal clefting sometimes present.
Pathological change
Possible diagnoses
Vacuolar change
Lupus erythematosus, dermatomyositis, drugs and poikiloderma
Interface-obscuring infiltrate
Erythema multiforme, fixed drug eruption, pityriasis lichenoides (acute), paraneoplastic pemphigus, lupus erythematosus (some)
Purpura
Lichenoid purpura
Cornoid lamella
Porokeratosis
Deep dermal infiltrate
Lupus erythematosus, syphilis, drugs, photolichenoid eruption
‘Satellite cell necrosis’
Graft-versus-host disease, eruption of lymphocyte recovery, erythema multiforme, paraneoplastic pemphigus, regressing plane warts, drug reactions
High apoptosis
Phototoxic reactions, adult-onset Still’s disease, acrokeratosis paraneoplastica
Prominent pigment incontinence
Poikiloderma, drugs, ‘racial pigmentation’ and an associated lichenoid reaction, erythema dyschromicum perstans and related entities
Eccrine duct involvement
Erythema multiforme (drug induced), lichen striatus, keratosis lichenoides chronica, periflexural exanthem of childhood
Additional pattern
Possible diagnoses
Spongiotic
Drug reactions (see spongiotic drug reactions), lichenoid contact dermatitis, lichen striatus, late-stage pityriasis rosea, superantigen ‘id’ reactions
Granulomatous
Lichen nitidus, lichen striatus (rare), lichenoid sarcoidosis, hepatobiliary disease, endocrinopathies, infective reactions including secondary syphilis, herpes zoster infection, HIV infection, tinea capitis, Mycobacterium marinum, and M. haemophilum; drug reactions (often in setting of Crohn’s disease or rheumatoid arthritis – atenolol, allopurinol, captopril, cimetidine, enalapril, hydroxychloroquine, simvastatin, sulfa drugs, tetracycline, diclofenac, erythropoietin)
Vasculitic
Pityriasis lichenoides, perniosis (some cases), pigmented purpuric dermatosis (lichenoid variant), persistent viral reactions, including herpes simplex
Vasculitic/spongiotic
Gianotti–Crosti syndrome, some other viral/putative viral diseases, rare drug reactions
LICHENOID (INTERFACE) DERMATOSES
LICHEN PLANUS
Treatment of lichen planus
Histopathology167
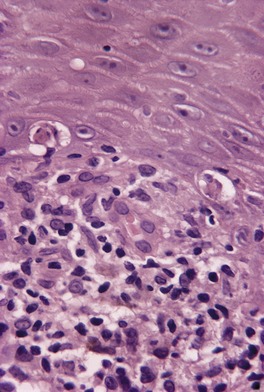
Fig. 3.2
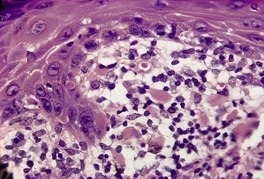
Fig. 3.3
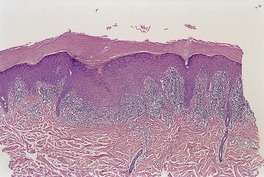
Fig. 3.4
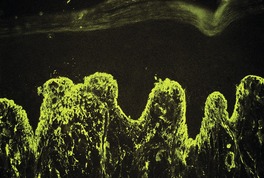
Fig. 3.5
Electron microscopy
LICHEN PLANUS VARIANTS
Atrophic lichen planus
Histopathology
Hypertrophic lichen planus
Histopathology
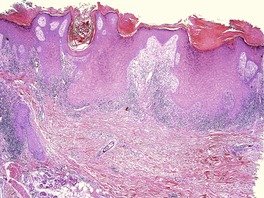
Fig. 3.6
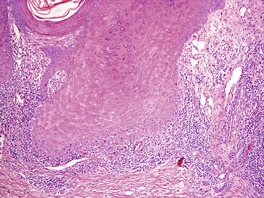
Fig. 3.7
Annular lichen planus
Linear lichen planus
Ulcerative (erosive) lichen planus
Histopathology
Oral lichen planus
Histopathology
Lichen planus erythematosus
Histopathology
Erythema dyschromicum perstans
Histopathology254
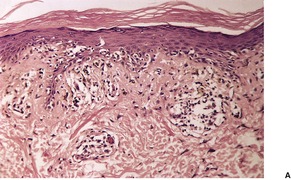
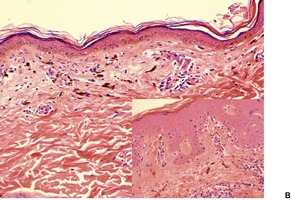
Fig. 3.8
Lichen planus actinicus
Histopathology287
Lichen planopilaris
Histopathology298.299. and 302.
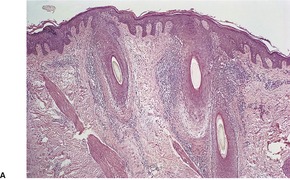
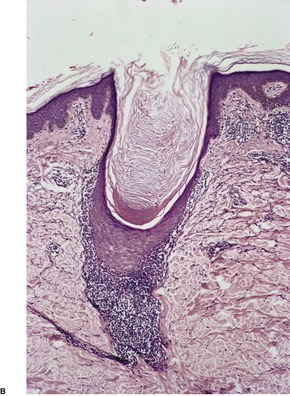
Fig. 3.9
Lichen planus pemphigoides
Histopathology
Electron microscopy
Keratosis lichenoides chronica
Histopathology
Lupus erythematosus–lichen planus overlap syndrome
LICHEN NITIDUS
Histopathology
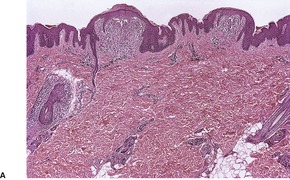
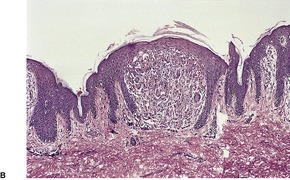
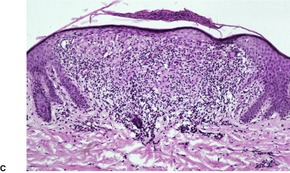
Fig. 3.10
Electron microscopy
LICHEN STRIATUS
Histopathology427.453.454. and 455.
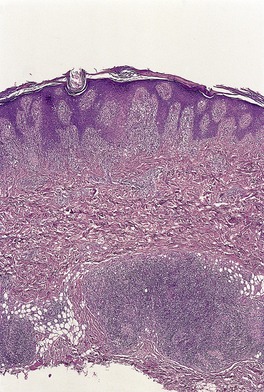
Fig. 3.11
Electron microscopy
LICHEN PLANUS-LIKE KERATOSIS (BENIGN LICHENOID KERATOSIS)
Histopathology462.463. and 474.
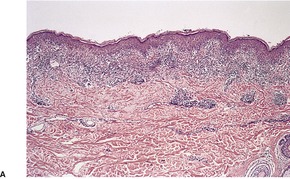
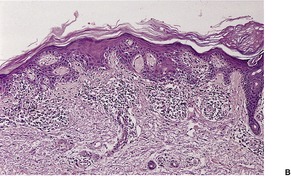
Fig. 3.12
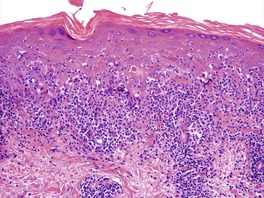
Fig. 3.13
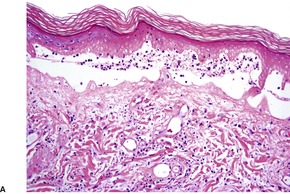
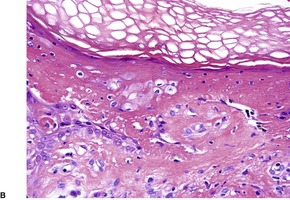
Fig. 3.14
The early/interface variant is more likely to be a reflection of the underlying lesion undergoing regression, rather than a variant sui generis.
Classic
Atrophic
Atypical (MF-like)
Bullous
TEN-like
‘Creeping’
Lupus stimulant
LICHENOID DRUG ERUPTIONS
Lichen planus pemphigoides: cinnarizine, ramipril
Lichenoid drug eruption: acetylsalicyclic acid, adalimumab, amlodipine, antimalarials, arsenicals, beta-blockers, captopril, carbamazepine, chlorpropamide, cyanamide, dactinomycin, dapsone, docetaxel, doxorubicin, enalapril, etanercept, ethambutol, glimepiride, gold, granulocyte colony-stimulating factor (local), hepatitis B vaccination, imatinib, indapamide, indoramin, infliximab, interferon alfa-2b, intravenous immunoglobulins, iodides, lansoprazole, mercury, metformin, methyldopa, naproxen, nicorandil, omeprazole, orlistat, pantoprazole, penicillamine, phenothiazine, pravastatin, quinidine, quinine, salsalate, simvastatin, spironolactone, streptomycin, suramin, terazosin, ticlopidine, tiopronin, valproic acid
Photolichenoid drug eruption: clopidogrel, nimesulide, pyrazinamide, solifenacin, sparfloxacin, tetracycline, thiazides, torsemide
Fixed drug eruption: acetaminophen (paracetamol), acetaminophen/indomethacin/granisetron/IL-2 combination, acetylsalicyclic acid, amoxicillin, amplodipine, antimalarials, antituberculous drugs, carbamazepine, celecoxib, cetirizine, chlormezanone, ciprofloxacin, clarithromycin, clioquinol, colchicine, cotrimoxazole, dextromethorphan, diltiazem, dimenhydrinate, diphenhydramine, enalapril, eperisone hydrochloride, erythromycin, etodolac, feprazone, fluconazole, fluoxetine, griseofulvin, ibuprofen, influenza vaccine, interferon-β, iomeprol, itraconazole, ketoconazole, lactose, lamotrigine, lansoprazole, levocetirizine, mefenamic acid, metamizole, metronidazole, minocycline, naproxen, nimesulide, nystatin, omeprazole, paclitaxel, penicillin, phenolphthalein, phenylbutazone, phenylpropanolamine hydrochloride, phenytoin, piroxicam, pseudoephedrine, quinine, rifampicin, S-carboxymethyl-L-cysteine, sulfamethoxazole, sulfonamides, tartrazene, temazepam, tenoxicam, terbinafine, tetracyclines, theophylline, ticlopidine, tinidazole, topotecan, tranexamic acid, tranquilizers, trimethoprim, tropisetron, vancomycin
Erythema multiforme/TEN: acarbose, acetaminophen (paracetamol), alfuzosin, allopurinol, amifostine, aminopenicillins, amphetamines, ampicillin/sulbactam, bezafibrate, bupropion, carbamazepine, ceftazidime, chloroquine, cimetidine, ciprofloxacin, citalopram, clarithromycin, clindamycin, clobazam, clonazepam, cocaine, colchicine, corticosteroid, cyclophosphamide, cytosine arabinoside, diacerein, doxycycline, escitalopram, ethambutol, etretinate, famotidine, fenoterol, fluoxetine, fluvoxamine, gemeprost, griseofulvin, hydroxychloroquine, imatinib, indapamide, indomethacin, iopentol, irbesartan, isoniazid, isoxicam, lamotrigine, lansoprazole, latanoprost eye drops, leflunomide, mefloquine, methotrexate, mifepristone, moxifloxacin, nevirapine, nitrogen mustard, nystatin, ofloxacin, oxaprozin, oxazepam, pantoprazole, paroxetine, phenylbutazone, phenytoin, piroxicam, progesterone, pseudoephedrine, ramipril, ranitidine, ritodrine, rituximab, rofecoxib, sennoside, sertraline, sorafenib, sulfamethoxazole, suramin, telithromycin, terbinafine, tetrazepam, thalidomide, theophylline, thiacetazone, ticlopidine, tramadol, trichloroethylene, trimethoprim, valdecoxib, valproic acid, vancomycin, voriconazole, zonisamide
Subacute lupus erythematosus: adalimumab, anastrozole, antihistamines, bupropion, calcium channel blockers, capecitabine, captopril, cilazapril, cinnarizine, diltiazem, efalizumab, etanercept, griseofulvin, infliximab, leflunomide, naproxen, nifedipine, oxyprenolol, phenytoin, piroxicam, ranitidine, simvastatin, terbinafine, thiazides, ticlopidine, tiotropium, verapamil
Systemic lupus erythematosus: allopurinol, atenolol, captopril, carbamazepine, chlorpromazine, clonidine, danazol, etanercept, ethosuximide, griseofulvin, hydralazine, hydrochlorothiazide, isoniazid, lithium, lovastatin, mesalazine, methimazole, methyldopa, minocycline, penicillin, penicillamine, phenobarbital, phenylbutazone, phenytoin, piroxicam, practolol, primidone, procainamide, propylthiouracil, quinidine, rifampicin, streptomycin, sulfonamides, terbinafine, tetracycline derivatives, thiamazole, trimethadione, valproate
Histopathology492. and 565.
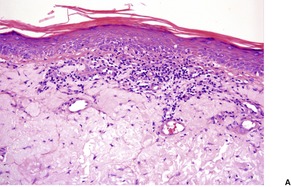
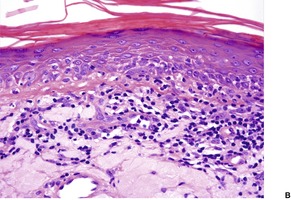
Fig. 3.15
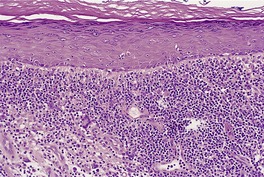
Fig. 3.16
FIXED DRUG ERUPTIONS
Histopathology573. and 685.
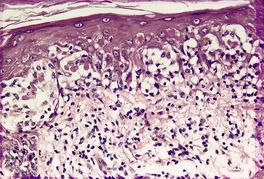
Fig. 3.17
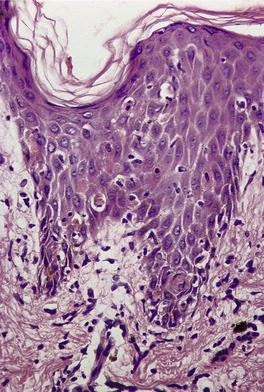
Fig. 3.18
Electron microscopy
ERYTHEMA MULTIFORME
Clinical variants
Etiology and pathogenesis
Histopathology690.835. and 836.
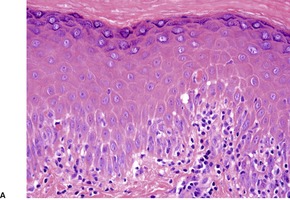
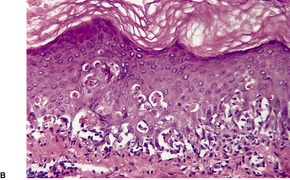
Fig. 3.19
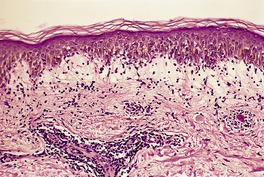
Fig. 3.20
Toxic epidermal necrolysis
Treatment of toxic epidermal necrolysis
Histopathology855. and 967.
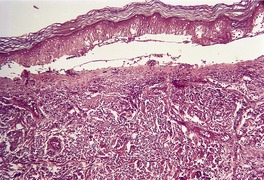
Fig. 3.21
GRAFT-VERSUS-HOST DISEASE
Histopathology978. and 1060.
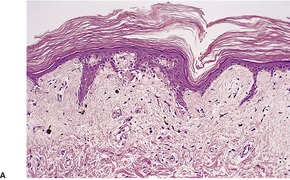
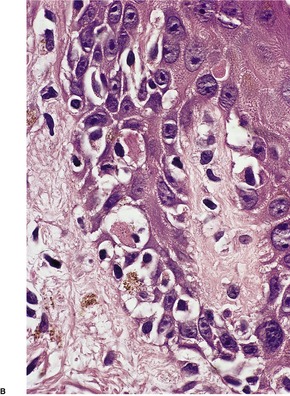
Fig. 3.22
Grade 0:
normal skin
Grade 1:
basal vacuolar change
Grade 2:
dyskeratotic cells in the epidermis and/or follicle, dermal lymphocytic infiltrate
Grade 3:
fusion of basilar vacuoles to form clefts and microvesicles
Grade 4:
separation of epidermis from dermis.
Electron microscopy
ERUPTION OF LYMPHOCYTE RECOVERY
Histopathology
AIDS INTERFACE DERMATITIS
LUPUS ERYTHEMATOSUS
Discoid lupus erythematosus
Treatment of discoid lupus erythematosus
Histopathology1097. and 1207.
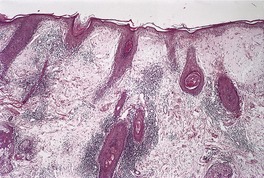
Fig. 3.23
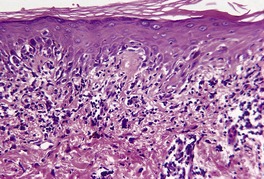
Fig. 3.24
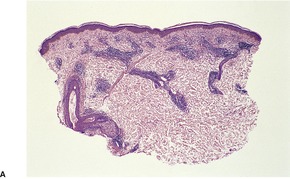
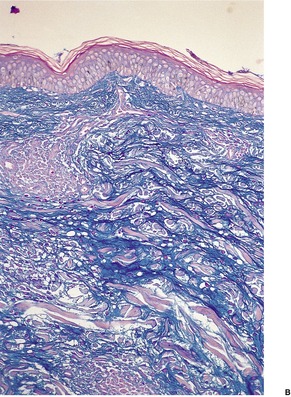
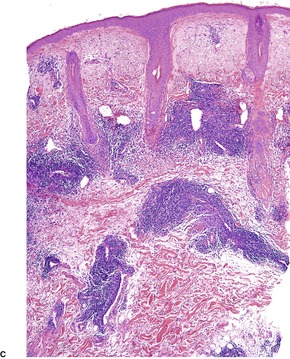
Fig. 3.25
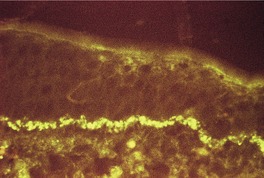
Fig. 3.26
Subacute lupus erythematosus
Treatment of subacute cutaneous lupus erythematosus
Histopathology1305
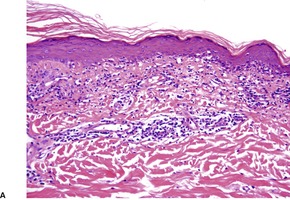
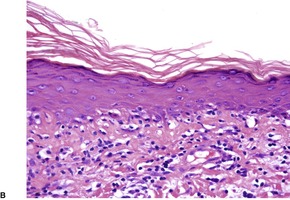
Fig. 3.27
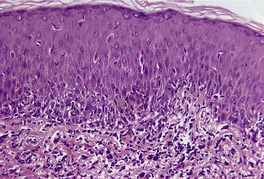
Fig. 3.28
Systemic lupus erythematosus
Investigations
Etiology
Treatment of systemic lupus erythematosus
Histopathology1097
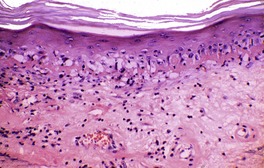
Fig. 3.29
LUPUS ERYTHEMATOSUS VARIANTS
Neonatal lupus erythematosus
Bullous lupus erythematosus
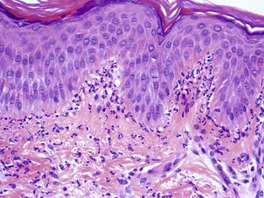
Fig. 3.30
Lupus panniculitis
DERMATOMYOSITIS
Treatment of dermatomyositis
Histopathology1689
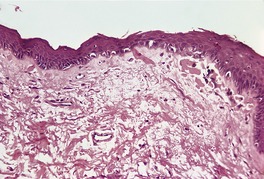
Fig. 3.31
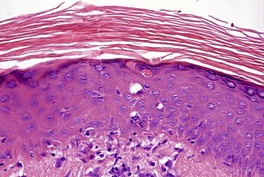
Fig. 3.32
POIKILODERMAS
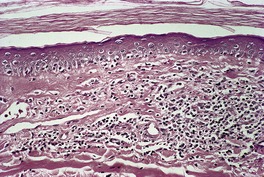
Fig. 3.33
POIKILODERMA CONGENITALE (ROTHMUND–THOMSON SYNDROME)
Histopathology
Hereditary sclerosing poikiloderma
KINDLER’S SYNDROME
Histopathology
CONGENITAL TELANGIECTATIC ERYTHEMA (BLOOM’S SYNDROME)
Histopathology
DYSKERATOSIS CONGENITA
OMIM
Inheritance
Gene defect
Gene product
Locus
Alternative name
305000
X-linked
DKC1
Dyskerin
Xq28
Zinsser–Cole–Engman syndrome
127550
AD
TERC
Telomerase RNA component
3q21–q28
Scoggins type
AD
TERT
Telomerase reverse transcriptase
5p15.33
AD
TINF2
?
14q11.2 (14q12?)
224230
AR
NOPIO (NOLA3)
?
15q14–q15
Nil
Histopathology
POIKILODERMA OF CIVATTE
Histopathology
OTHER LICHENOID (INTERFACE) DISEASES
LICHEN SCLEROSUS ET ATROPHICUS
PITYRIASIS LICHENOIDES
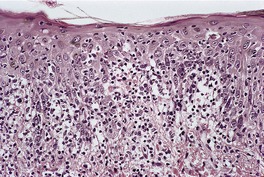
Fig. 3.34
PERSISTENT VIRAL REACTIONS
PERNIOSIS
PARANEOPLASTIC PEMPHIGUS
LICHENOID PURPURA
LICHENOID CONTACT DERMATITIS
STILL’S DISEASE (ADULT ONSET)
LATE SECONDARY SYPHILIS
POROKERATOSIS
DRUG ERUPTIONS
PHOTOTOXIC DERMATITIS
PRURIGO PIGMENTOSA
ERYTHRODERMA
MYCOSIS FUNGOIDES
REGRESSING WARTS AND TUMORS
LICHEN AMYLOIDOSUS
VITILIGO
LICHENOID TATTOO REACTION
MISCELLANEOUS CONDITIONS
LICHENOID AND GRANULOMATOUS DERMATITIS
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The lichenoid reaction pattern (‘interface dermatitis’)
Lichenoid (interface) dermatoses38
Lichen planus38
Lichen planus variants40
Atrophic lichen planus40
Hypertrophic lichen planus40
Annular lichen planus41
Linear lichen planus41
Ulcerative (erosive) lichen planus41
Oral lichen planus41
Lichen planus erythematosus42
Erythema dyschromicum perstans42
Lichen planus actinicus43
Lichen planopilaris43
Lichen planus pemphigoides43
Keratosis lichenoides chronica44
Lupus erythematosus–lichen planus overlap syndrome45
Lichen nitidus45
Lichen striatus45
Lichen planus-like keratosis (benign lichenoid keratosis)47
Lichenoid drug eruptions48
Fixed drug eruptions50
Graft-versus-host disease55
Eruption of lymphocyte recovery57
AIDS interface dermatitis57
Dermatomyositis64
Other lichenoid (interface) diseases68
Lichen sclerosus et atrophicus69
Pityriasis lichenoides69
Persistent viral reactions69
Perniosis69
Paraneoplastic pemphigus69
Lichenoid purpura69
Lichenoid contact dermatitis69
Still’s disease (adult onset)69
Late secondary syphilis69
Porokeratosis69
Drug eruptions70
Phototoxic dermatitis70
Prurigo pigmentosa70
Erythroderma70
Mycosis fungoides70
Regressing warts and tumors70
Lichen amyloidosus70
Vitiligo70
Lichenoid tattoo reaction70
Miscellaneous conditions70
Lichenoid and granulomatous dermatitis70
The lichenoid reaction pattern (lichenoid tissue reaction, interface dermatitis) is characterized histologically by epidermal basal cell damage.1.2. and 3. This takes the form of cell death and/or vacuolar change (liquefaction degeneration). The cell death usually involves only scattered cells in the basal layer which become shrunken with eosinophilic cytoplasm. These cells, which have been called Civatte bodies, often contain pyknotic nuclear remnants. Sometimes, fine focusing up and down will reveal smaller cell fragments, often without nuclear remnants, adjacent to the more obvious Civatte bodies. 4 These smaller fragments have separated from the larger bodies during the process of cell death. Ultrastructural studies have shown that the basal cells in the lichenoid reaction pattern usually die by apoptosis, a comparatively recently described form of cell death, which is quite distinct morphologically from necrosis.5. and 6.
Before discussing the features of apoptosis, mention will be made of the term ‘interface dermatitis’, which is widely used. It has been defined as a dermatosis in which the infiltrate (usually composed mostly of lymphocytes) appears ‘to obscure the junction when sections are observed at scanning magnification’. 7 The term is not used uniformly or consistently. Some apply it to most dermatoses with the lichenoid tissue reaction. Others use it for the subgroup in which the infiltrate truly obscures the interface (erythema multiforme, fixed drug eruption, paraneoplastic pemphigus, some cases of subacute lupus erythematosus and pityriasis lichenoides). The infiltrate may obscure the interface in lymphomatoid papulosis, but basal cell damage is not invariable. Many apply the term, also, to lichen planus and variants, in which the infiltrate characteristically ‘hugs’ the basal layer without much extension into the epidermis beyond the basal layer. Crowson et al have expanded the concept of interface dermatitis to include neutrophilic and lymphohistiocytic forms, in addition to the traditional lymphocytic type. They also subdivide the lymphocytic type into a cell-poor type and a cell-rich type. 8 Erythema multiforme, which they list as a cell-poor variant, is sometimes quite ‘cell rich’. The author prefers the traditional term ‘lichenoid’ for this group of dermatoses because it is applicable more consistently than interface dermatitis and it is less likely to be applied as a ‘final sign-out diagnosis’, which is often the case with the term interface dermatitis. The term is so entrenched that it is unlikely to disappear from the lexicon of dermatopathology.
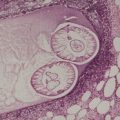
In apoptosis, single cells become condensed and then fragment into small bodies by an active budding process (Fig. 3.1). In the skin, these condensed apoptotic bodies are known as Civatte bodies (see above). The smaller apoptotic bodies, some of which are beyond the resolution of the light microscope, are usually phagocytosed quickly by adjacent parenchymal cells or by tissue macrophages. 5 Cell membranes and organelles remain intact for some time in apoptosis, in contradistinction to necrosis where breakdown of these structures is an integral and prominent part of the process. Keratinocytes contain tonofilaments which act as a ‘straitjacket’ within the cell, and therefore budding and fragmentation are less complete in the skin than they are in other cells in the body undergoing death by apoptosis. This is particularly so if the keratinocyte has accumulated filaments in its cytoplasm, as occurs with its progressive maturation in the epidermis. The term ‘dyskeratotic cell’ is usually used for these degenerate keratinocytes. The apoptotic bodies that are rich in tonofilaments are usually larger than the others; they tend to ‘resist’ phagocytosis by parenchymal cells, although some are phagocytosed by macrophages. Others are extruded into the papillary dermis, where they are known as colloid bodies. These bodies appear to trap immunoglobulins non-specifically, particularly the IgM molecule, which is larger than the others. Apoptotic cells can be labeled by the TUNEL reaction. 9
(A) Apoptosis of a basal keratinocyte in lichen planus. There is surface budding and some redistribution of organelles within the cytoplasm. Electron micrograph ×12 000. (B) A tiny budding fragment in which the mitochondria have intact cristae. (×25 000)
Some of the diseases included within the lichenoid reaction pattern show necrosis of the epidermis rather than apoptosis; in others, the cells have accumulated so many cytoplasmic filaments prior to death that the actual mechanism – apoptosis or necrosis – cannot be discerned by light or electron microscopy. The term ‘filamentous degeneration’ has been suggested for these cells; 10 on light microscopy, they are referred to as ‘dyskeratotic cells’ (see above). Some dermatopathologists use the term ‘necrotic keratinocyte’ for these cells and, also, for keratinocytes that are obviously apoptotic. It should be noted that apoptotic keratinocytes have been seen in normal skin, indicating that cell deletion also occurs as a normal physiological phenomenon.11.12. and 13. As Afford and Randhawa have so eloquently stated ‘Apoptosis is the genetically regulated form of cell death that permits the safe disposal of cells at the point in time when they have fulfilled their intended biological function.’14 It also plays a role in the elimination of the inflammatory infiltrate at the end stages of wound healing. 15
Although it is beyond the scope of this book, readers interested in apoptosis and the intricate mechanisms of its control should read the excellent studies published on this topic.16.17.18.19.20.21.22.23. and 24. The various ‘death receptors’, essential effectors of any programmed cell death, were reviewed in 2003. 25 An important member of this group is tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) which preferentially induces apoptosis in transformed but not normal cells. It is expressed in normal skin and cutaneous inflammatory diseases. 26 Another cell component that plays a role in apoptosis is the mitochondrion. This topic was reviewed in 2006. 27
Ackerman has continued to present a minority view that apoptosis is a type of necrosis. 28 In reality, each is a distinctive form of cell death.
Vacuolar change (liquefaction degeneration) is often an integral part of the basal damage in the lichenoid reaction. Sometimes it is more prominent than the cell death. It results from intracellular vacuole formation and edema, as well as from separation of the lamina densa from the plasma membrane of the basal cells. Vacuolar change is usually prominent in lupus erythematosus, particularly the acute systemic form, and in dermatomyositis and some drug reactions.
As a consequence of the basal cell damage, there is variable melanin incontinence resulting from interference with melanin transfer from melanocytes to keratinocytes, as well as from the death of cells in the basal layer. 1 Melanin incontinence is particularly prominent in some drug-induced and solar-related lichenoid lesions, as well as in patients with marked racial pigmentation.
Another feature of the lichenoid reaction pattern is a variable inflammatory cell infiltrate. This varies in composition, density and distribution according to the disease. An assessment of these characteristics is important in distinguishing the various lichenoid dermatoses. As apoptosis, unlike necrosis, does not itself evoke an inflammatory response, it can be surmised that the infiltrate in those diseases with prominent apoptosis is of pathogenetic significance and not a secondary event. 5 Furthermore, apoptosis is the usual method of cell death resulting from cell-mediated mechanisms, whereas necrosis and possibly vacuolar change result from humoral factors, including the deposition of immune complexes.
One study has given some insight into the possible mechanisms involved in the variability of expression of the lichenoid tissue reaction in several of the diseases within this group. The study examined the patterns of expression of the intercellular adhesion molecule-1 (ICAM-1). 29 Keratinocytes in normal epidermis have a low constitutive expression of ICAM-1, rendering the normal epidermis resistant to interaction with leukocytes. Therefore, induction of ICAM-1 expression may be an important factor in the induction of leukocyte-dependent damage to keratinocytes. 29 In lichen planus, ICAM-1 expression is limited to basal keratinocytes, while in subacute cutaneous lupus erythematosus there is diffuse epidermal ICAM-1 expression, sometimes with basal accentuation. This pattern is induced by ultraviolet radiation and possibly mediated by tumor necrosis factor-α. In erythema multiforme, there is strong basal expression of ICAM-1, with cell surface accentuation and similar pockets of suprabasal expression, probably induced by herpes simplex virus infection. 29
In summary, the lichenoid reaction pattern includes a heterogeneous group of diseases which have in common basal cell damage. 30 The histogenesis is also diverse and includes cell-mediated and humoral immune reactions and possibly ischemia in one condition. A discussion of the mechanisms involved in producing apoptosis is included in several of the diseases that follow. Scattered apoptotic keratinocytes can also be seen in the sunburn reaction in response to ultraviolet radiation;31. and 32. these cells are known as ‘sunburn cells’. 33 A specific histological diagnosis can usually be made by attention to such factors as:
A discussion of the various lichenoid (interface) dermatoses follows. The conditions listed as ‘other lichenoid (interface) diseases’ are discussed only briefly as they are considered in detail in other chapters.
Lichen planus, a relatively common eruption of unknown etiology, displays violaceous, flat-topped papules, which are usually pruritic.35. and 36. A network of fine white lines (Wickham’s striae) may be seen on the surface of the papules. There is a predilection for the flexor surface of the wrists, the trunk, the thighs, and the genitalia. Palmoplantar lichen planus appears to be more common than once thought.37. and 38. It is one of the most disabling, painful, and therapy-resistant variants of lichen planus. 39 Oral lesions are common; rarely, the esophagus is also involved.40. and 41. Lesions localized to the lip, 42 vulva,43. and 44. and to an eyelid45 have been reported. Lichen planus localized to a radiation field may represent an isomorphic response.46. and 47. It has also developed in a healed herpes zoster scar. 48 Nail changes occur49.50.51. and 52. and, as with oral lesions, these may be the only manifestations of the disease.53. and 54. Clinical variants include atrophic, annular, hypertrophic, linear, zosteriform,55.56. and 57. erosive, oral, actinic, follicular, erythematous, and bullous variants. They are discussed further below. An eruptive variant also occurs. 58 Spontaneous resolution of lichen planus is usual within 12 months, although postinflammatory pigmentation may persist for some time afterwards. 59
Familial cases are uncommon, and rarely these are associated with HLA-D7.60.61.62. and 63. An association with HLA-DR1 has been found in non-familial cases. 64 There is an increased frequency of HLA-DR6 in Italian patients with hepatitis C virus-associated oral lichen planus. 65 Lichen planus is rare in children,66.67.68.69.70.71.72. and 73. but some large series have been published.74.75. and 76. Lichen planus has been reported in association with immunodeficiency states, 77 internal malignancy,78. and 79. including thymoma, 80 primary biliary cirrhosis,81. and 82. peptic ulcer (but not Helicobacter pylori infection), 83 chronic hepatitis C infection,84.85.86.87.88.89.90.91.92.93. and 94. hepatitis B vaccination,95.96.97.98.99.100.101.102.103. and 104. herpesvirus type 7 (HHV-7) replication, 105 simultaneous measles-mumps-rubella and diphtheria-tetanus-pertussis-polio vaccinations, 106 stress, 107 vitiligo, 108 pemphigus, 109 porphyria cutanea tarda, 110 radiotherapy, 111 ulcerative colitis, 112 chronic giardiasis, 113 a Becker’s nevus, 114 and lichen sclerosus et atrophicus with coexisting morphea. 115 Despite the association between lichen planus and hepatitis C (HCV) infection, its incidence in patients with lichen planus in some parts of the world is not increased when compared with a control group.116. and 117. The exacerbation or appearance of lichen planus during the treatment of HCV infection and other diseases with alpha-interferon has been reported. 118 Furthermore effective therapy for the HCV does not clear the lichen planus. 119 Reports linking lichen planus to infection with human papillomavirus may be a false-positive result.120. and 121. Squamous cell carcinoma is a rare complication of the oral and vulval cases of lichen planus and of the hypertrophic and ulcerative variants (see below).122.123.124.125. and 126. A contact allergy to metals, flavorings and plastics may be important in the etiology of oral lichen planus. 127 The role of mercury in dental amalgams is discussed further below.
Cell-mediated immune reactions appear to be important in the pathogenesis of lichen planus. 128 It has been suggested that these reactions are precipitated by an alteration in the antigenicity of epidermal keratinocytes, possibly caused by a virus or a drug or by an allogeneic cell. 129 Keratinocytes in lichen planus express HLA-DR on their surface and this may be one of the antigens which has an inductive or perpetuating role in the process.130.131. and 132. Keratinocytes also express fetal cytokeratins (CK13 and CK8/18) but whether they are responsible for triggering the T-cell response is speculative. 133 The cellular response, initially, consists of CD4+ lymphocytes; 134 they are also increased in the peripheral blood. 135 In recent years attention has focused on the role of cytotoxic CD8+ lymphocytes in a number of cell-mediated immune reactions in the skin. They appear to play a significant role as the effector cell, whereas the CD4+ lymphocyte, usually present in greater numbers, 136 plays its traditional ‘helper’ role. In lichen planus, CD8+ cells appear to recognize an antigen associated with MHC class I on lesional keratinocytes, resulting in their death by apoptosis. 137Bcl-2, a proto-oncogene that protects cells from apoptosis, is increased in lichen planus. 138 It may allow some cells to escape apoptosis, prolonging the inflammatory process. 138 The recruitment of lymphocytes to the interface region may be the result of the chemokine MIG (monokine induced by interferon-γ). 139 Lymphokines produced by these T lymphocytes, including interferon-γ, interleukins-1β, -4 and -6, perforin, 140 granzyme B, 141 granulysin, 142 T-cell-restricted intracellular antigen (Tia-1), and tumor necrosis factor, may have an effector role in producing the apoptosis of keratinocytes.143. and 144. The other pathway involves the binding of Fas ligand to Fas, which triggers a caspase cascade.145. and 146. Gene expression profiling in lichen planus has found that type I IFN inducible genes are significantly expressed. 147 Plasmacytoid dendritic cells appear to be a major source of these type I interferons in lichen planus. They play a major role in cytotoxic skin inflammation by increasing the expression of IPIO/CXCRIO and recruiting effector cells via CXCR3. 148 The CXCR3 ligand, CXCL9, is the most significant marker for lichen planus. 147 A unique subclass of cytotoxic T lymphocyte (γδ) is also found in established lesions. 149 Langerhans cells are increased and it has been suggested that these cells initially process the foreign antigen. 131 Factor XIIIa-positive cells and macrophages expressing lysozyme are found in the dermis. 134
Increased oxidative stress, increased lipid peroxidation, and an imbalance in the antioxidant defense system are present, but their exact role in the pathogenesis of lichen planus is unknown. 150
Matrix metalloproteinases may play a concurrent role by destroying the basement membrane. 151 Evidence from an animal model suggests that keratinocytes require cell survival signals, derived from the basement membrane, to prevent the onset of apoptosis. 152 In oral lichen planus, MMP-1 and MMP-3 may be principally associated with erosion development. 153 Altered levels of heat shock proteins are found in the epidermis in lichen planus. 154
Most studies have found no autoantibodies and no alteration in serum immunoglobulins in lichen planus. 155 However, a lichen planus-specific antigen has been detected in the epidermis and a circulating antibody to it has been found in the serum of individuals with lichen planus.156. and 157. Its pathogenetic significance remains uncertain. Antibodies to desmoplakins I and II have been found in oral and genital lesions, possibly representing epitope spreading. 158
Replacement of the damaged basal cells is achieved by an increase in actively dividing keratinocytes in both the epidermis and the skin appendages. This is reflected in the pattern of keratin expression, which resembles that seen in wound healing; cytokeratin 17 (CK17) is found in suprabasal keratinocytes. 159
Potent topical corticosteroids remain the treatment of choice for lichen planus in patients with classic and localized disease. For widespread disease and mucosal lesions, a short course of systemic corticosteroids may provide some relief. 160 Cyclosporine (ciclosporin), hydroxychloroquine, retinoids, dapsone, mycophenolate mofetil, 161 sulfasalazine, 162 alefacept, 163 and efalizumab164 have all been used at various times. Erosive oral disease has been treated with tacrolimus mouthwash, 165 while erosive flexural lichen planus has responded to thalidomide and 0.1% tacrolimus ointment. 166 Palmoplantar disease may be resistant to treatment and require cyclosporine. 38
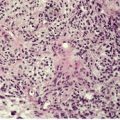
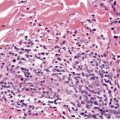
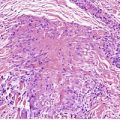
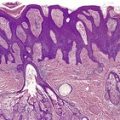
The basal cell damage in lichen planus takes the form of multiple, scattered Civatte bodies (Fig. 3.2). Eosinophilic colloid bodies, which are PAS positive and diastase resistant, are found in the papillary dermis (Fig. 3.3). They measure approximately 20 µm in diameter. The basal damage is associated with a band-like infiltrate of lymphocytes and some macrophages which press against the undersurface of the epidermis (Fig. 3.4). Occasional lymphocytes extend into the basal layer, where they may be found in close contact with basal cells and sometimes with Civatte bodies. The infiltrate does not obscure the interface or extend into the mid-epidermis, as in erythema multiforme and fixed drug eruptions. Karyorrhexis is sometimes seen in the dermal infiltrate. 168 Rarely, plasma cells are prominent in the cutaneous lesions;169.170.171. and 172. they are invariably present in lesions adjacent to or on mucous membranes. There is variable melanin incontinence but this is most conspicuous in lesions of long duration and in dark-skinned people.
Lichen planus. Two apoptotic keratinocytes (Civatte bodies) are present in the basal layer of the epidermis. An infiltrate of lymphocytes touches the undersurface of the epidermis. (H & E)
Lichen planus. There are numerous colloid bodies in the papillary dermis. (H & E)
Lichen planus. A band-like infiltrate of lymphocytes fills the papillary dermis and touches the undersurface of the epidermis. (H & E)
Other characteristic epidermal changes include hyperkeratosis, wedge-shaped areas of hypergranulosis related to the acrosyringia and acrotrichia, and variable acanthosis. At times the rete ridges become pointed, imparting a ‘saw tooth’ appearance to the lower epidermis. There is sometimes mild hypereosinophilia of keratinocytes in the malpighian layer. Small clefts (Caspary–Joseph spaces) 173 may form at the dermoepidermal junction secondary to the basal damage. The eccrine duct adjacent to the acrosyringium is sometimes involved. 174 A variant in which the lichenoid changes were localized entirely to the acrosyringium has been reported. 175 Transepidermal elimination with perforation is another rare finding. 176 The formation of milia may be a late complication. 177
Ragaz and Ackerman have studied the evolution of lesions in lichen planus. 167 They found an increased number of Langerhans cells in the epidermis in the very earliest lesions, before there was any significant infiltrate of inflammatory cells in the dermis. In resolving lesions, the infiltrate is less dense and there may be minimal extension of the inflammatory infiltrate into the reticular dermis.
As already mentioned, some diseases exhibiting the lichenoid tissue reaction may also show features of another tissue reaction pattern as a major or minor feature. These conditions are listed in Table 3.3.
Direct immunofluorescence of involved skin shows colloid bodies in the papillary dermis, staining for complement and immunoglobulins, particularly IgM. An irregular band of fibrin is present along the basal layer in most cases. Often there is irregular extension of the fibrin into the underlying papillary dermis (Fig. 3.5). A recent study has found colloid bodies in 60% of cases of lichen planus, while fibrin was present in all cases. 178 Immunofluorescent analysis of the basement membrane zone, using a range of antibodies, suggests that disruption occurs in the lamina lucida region. 179 Other studies have shown a disturbance in the epithelial anchoring system. 180
Lichen planus. A band of fibrin involves the basement membrane zone and extends into the papillary dermis. (Direct immunofluorescence)
Ultrastructural studies have confirmed that lymphocytes attach to basal keratinocytes, resulting in their death by apoptosis.4.5. and 181. Many cell fragments, beyond the limit of resolution of the light microscope, are formed during the budding of the dying cells. The cell fragments are phagocytosed by adjacent keratinocytes and macrophages. 182 The large tonofilament-rich bodies that result from redistribution of tonofilaments during cell fragmentation appear to resist phagocytosis and are extruded into the upper dermis, where they are recognized on light microscopy as colloid bodies. 183 Various studies have confirmed the epidermal origin of these colloid bodies.184. and 185. There is a suggestion from some experimental work that sublethal injury to keratinocytes may lead to the accumulation of tonofilaments in their cytoplasm. Some apoptotic bodies contain more filaments than would be accounted for by a simple redistribution of the usual tonofilament content of the cell.
A number of clinical variants of lichen planus occur. In some, typical lesions of lichen planus are also present. These variants are discussed in further detail below.
Atrophic lesions may resemble porokeratosis clinically. Typical papules of lichen planus are usually present at the margins. A rare form of atrophic lichen planus is composed of annular lesions.186.187.188.189. and 190. It is composed of violaceous plaques of annular morphology with central atrophy. 191 Hypertrophic lichen planus has been reported at the edge of a plaque of annular atrophic lichen planus. 192 Experimentally, there is an impaired capacity of the atrophic epithelium to maintain a regenerative steady state.
The epidermis is thin and there is loss of the normal rete ridge pattern. The infiltrate is usually less dense than in typical lichen planus. It may be lost in the center of the lesions.
Hypertrophic lesions are usually confined to the shins, although sometimes they are more generalized. They appear as single or multiple pruritic plaques, which may have a verrucous appearance; 193 they usually persist for many years. Rarely, squamous cell carcinoma develops in lesions of long standing.194.195.196. and 197. Cutaneous horns, keratoacanthoma, and verrucous carcinoma may also develop in hypertrophic lichen planus.198.199. and 200.
Hypertrophic lichen planus has been reported in several patients infected with the human immunodeficiency virus. 201 It also occurs in patients with HCV infection. 202 It may occur in children. 203
The epidermis shows prominent hyperplasia and overlying orthokeratosis (Fig. 3.6). At the margins there is usually psoriasiform hyperplasia representing concomitant changes of lichen simplex chronicus secondary to the rubbing and scratching. If the epidermal hyperplasia is severe it may mimic a squamous cell carcinoma on a shave biopsy. 204 Vertically oriented collagen (‘vertical-streaked collagen’) is present in the papillary dermis in association with the changes of lichen simplex chronicus.
Hypertrophic lichen planus. The epidermis shows irregular hyperplasia. The dermal infiltrate is concentrated near the tips of the rete ridges. (H & E)
The basal cell damage is usually confined to the tips of the rete ridges and may be missed on casual observation (Fig. 3.7). The infiltrate is not as dense or as band-like as in the usual lesions of lichen planus. A few eosinophils and plasma cells may be seen in some cases in which the ingestion of beta-blockers can sometimes be incriminated.
Hypertrophic lichen planus. There are a number of Civatte bodies near the tips of the rete ridges. (H & E)
Xanthoma cells have been found in the dermis, localized to a plaque of hypertrophic lichen planus, in a patient with secondary hyperlipidemia. 205 This is an example of dystrophic xanthomatization.
Annular lichen planus is one of the rarer clinical forms of lichen planus. In a series of 20 patients, published some years ago, 18 were men and 2 women. Sites of involvement included the axilla, penis, extremities, and groin. 206 Eighteen of the patients had purely annular lesions, whereas two of the patients had a few purple polygonal papules as well. The majority of lesions showed central clearing with a purple to white annular edge. Lesions varied from 0.5 to 2.5 cm in diameter. Atrophic annular lesions were discussed with atrophic lichen planus (see p. 40). The majority of patients were asymptomatic. Oral and genital lesions have been reported in annular lichen planus. 207
The cases reported as annular lichenoid dermatitis of youth appear to be a distinct entity, but further reports will be necessary to clarify its exact position in the spectrum of lichen planus. 208 The lesions are persistent erythematous macules and annular patches mostly localized on the groin and flanks. In all cases the clinical picture has been suggestive of morphea, mycosis fungoides, or annular erythema but these conditions could be excluded on the basis of the distinctive superficial lichenoid reaction with massive necrosis/apoptosis of the keratinocytes at the tips of the rete ridges. 208 Patch testing has given negative results. 209
Linear lichen planus is a rare variant which must be distinguished from linear nevi and other dermatoses with linear variants.210. and 211. It occurs in less than 0.5% of patients with lichen planus. 212 Linear lichen planus usually involves the limbs. It may follow the lines of Blaschko.213. and 214. It has been reported in association with hepatitis C infection, 212 HIV infection, 215 and metastatic carcinoma. 216
Sometimes linear lesions are associated with disseminated non-segmental papules of ordinary lichen planus. The linear lesions are usually more pronounced in these combined cases. 217
Ulcerative lichen planus (erosive lichen planus) is characterized by ulcerated and bullous lesions on the feet.218. and 219. Mucosal lesions, alopecia, and more typical lesions of lichen planus are sometimes present. Squamous cell carcinoma may develop in lesions of long standing. Variants of ulcerative lichen planus involving the perineal region, 220 penis, 221 the mouth, 222 or the vulva, vagina and mouth – the vulvovaginal-gingival syndrome43.223.224.225. and 226. – have been reported. A patient with erosive lesions of the flexures has been reported. 227
Castleman’s tumor (giant lymph node hyperplasia) and malignant lymphoma are rare associations of erosive lichen planus;228. and 229. long-term therapy with hydroxyurea and infection with hepatitis C are others.230. and 231. Screening for hepatitis C and B has not been considered necessary for vulval lichen planus in some countries. 232
Antibodies directed against a nuclear antigen of epithelial cells have been reported in patients with erosive lichen planus of the oral mucosa. 233 Weak circulating basement membrane zone antibodies are also present. 234
High potency topical corticosteroids have been used to treat erosive lichen planus. Relief of symptoms was obtained in 71% of cases of vulvar disease in one series. 235 A good response to topical tacrolimus, particularly in vulvar disease, has been achieved in recent years.236.237.238. and 239. Azathioprine, retinoids, dapsone, methotrexate, and hydroxychloroquine have also been used, but there have been no controlled trials of these various treatments.232. and 240. Photodynamic therapy can also be used.
Penile erosive lichen planus responded to circumcision in one case. 241
There is epidermal ulceration with more typical changes of lichen planus at the margins of the ulcer. Plasma cells are invariably present in cases involving mucosal surfaces. Eosinophils were prominent in the oral lesions of a case associated with methyldopa therapy. In erosive lichen planus of the vulva there is widespread disruption in several basement membrane zone components including hemidesmosomes and anchoring fibrils. 242
Oral lichen planus has a prevalence of about 0.5% to 2%. It is a disease of middle-aged and older persons, with a female predominance. 243 The disease may persist for many years despite treatment. Spontaneous remission is rare. 244
There is a low prevalence of oral lichen planus among hepatitis C (HCV) infected patients;245.246. and 247. the keratotic form of oral lichen planus is more prevalent in this disease. 202
There has been a resurgence of interest in the role of an allergy to mercury in dental amalgams in the pathogenesis of oral lichen planus. Dental plaque and calculus, which have also been shown to contain mercury, are also associated with the disease. 248 It appears that in cases unassociated with cutaneous lichen planus, oral lichen planus may often be cleared by the partial or complete removal of amalgam fillings, if there is a positive patch test reaction to mercury compounds.243. and 249. Because mercury-associated disease does not have all the clinical and/or histological features of oral lichen planus, the term ‘oral lichenoid lesion’ is sometimes used for these cases. 247 In one case lichen planus developed in a herpes zoster scar on the face following an amalgam (mercury) filling. 250 Oral squamous cell carcinoma is a rare complication of oral lichen planus. 244 It appears that desmocollin-1 expression in oral atrophic lichen planus is a powerful predictor of the development of dysplasia, while desmocollin-1 and E-cadherin expression are predictors of the development of cancer. 251
Oral lichen planus mimics to varying degrees the changes seen in cutaneous disease. The infiltrate is usually quite heavy, and it may contain plasma cells, particularly in erosive forms when neutrophils may also be present. Both cells are also found in amalgam-associated disease. Apoptotic keratinocytes tend to occur at a slightly higher level in the mucosa than they do in the cutaneous form, possibly a reflection of amalgam-related cases. Features said to be more likely in amalgam-associated disease are: deep extension of the infiltrate, perivascular extension of the infiltrate, and the presence of plasma cells and neutrophils in the connective tissue. 252
Lichen planus erythematosus has been challenged as an entity. Non-pruritic, red papules, with a predilection for the forearms, have been described. 193
A proliferation of blood vessels may be seen in the upper dermis in addition to the usual features of lichen planus.
Erythema dyschromicum perstans (ashy dermatosis, lichen planus pigmentosus) 253 is a slowly progressive, asymptomatic, ash-colored or brown macular hyperpigmentation254.255. and 256. which has been reported from most parts of the world; it is most prevalent in Latin America. 257 Lesions are often quite widespread, although there is a predilection for the trunk and upper limbs. Unilateral258 and linear lesions259 have been described. Periorbital hyperpigmentation is a rare presentation of this disease. 260 Activity of the disease may cease after several years. Resolution is more likely in children than in adults.261. and 262. Recently, it has been proposed that ‘erythema dyschromicum perstans’ should be used when lesions have, or have previously had, an erythematous border, while ‘ashy dermatosis’ should be used for other cases without this feature. 263 Most clinicians regard the terms as synonymous.
Erythema dyschromicum perstans has been regarded as a macular variant of lichen planus264 on the basis of the simultaneous occurrence of both conditions in several patients256.265. and 266. and similar immunopathological findings.267. and 268. Both paraphenylenediamine, and aminopenicillins have been incriminated in its etiology,261. and 264. although this has not been confirmed. This condition has also been reported in patients with HIV infection, 269 and with HCV infection. 270 There appears to be a genetic susceptibility to the disease. In one Mexican study, there was a significant increase in HLA-DR4, particularly the *0407 subtype, in patients with the disease. 257
Lichen planus pigmentosus, originally reported from India, is thought by some to be the same condition,255. and 264. although this has been disputed.263.271.272. and 273. A linear variant has been reported. 274 In a study of 124 patients with lichen planus pigmentosus from India, the face and neck were the commonest sites affected with pigmentation varying from slate gray to brownish-black. 275 Lichen planus was also present in 19 patients. 275 The term ‘lichen planus pigmentosus inversus’ has been used for cases with predominant localization of the disease in intertriginous areas.276.277. and 278. Lichen planus pigmentosus has been reported in association with a head and neck cancer and with concurrent acrokeratosis paraneoplastica (see p. 506). Both conditions cleared after treatment of the cancer. 279
Various therapies have been tried for erythema dyschromicum perstans, but with little benefit. They include sun protection, chemical peels, corticosteroids, and chloroquine. 280 Some patients have responded to dapsone, 280 and to clofazimine. 281
In the active phase, there is a lichenoid tissue reaction with basal vacuolar change and occasional Civatte bodies (Fig. 3.8). The infiltrate is usually quite mild in comparison to lichen planus. Furthermore, there may be deeper extension of the infiltrate, which is usually perivascular. There may also be mild exocytosis of lymphocytes. 273 There is prominent melanin incontinence and this is the only significant feature in older lesions. Subepidermal fibrosis was present in one case. 282 The pigment usually extends deeper in the dermis than in postinflammatory pigmentation of other causes. 271 Cases reported as lichen planus pigmentosus (see above) have similar histological features. 272
Erythema dyschromicum perstans. (A) There is patchy basal cell damage and some pigment incontinence. (B) Another case of ‘ashy dermatosis’ which has almost ‘burnt-out’. There is marked melanin incontinence. (H & E)
Immunofluorescence has shown IgM, IgG, and complement-containing colloid bodies in the dermis, as in lichen planus. There was a predominance of CD8+ lymphocytes in the dermis in one study. 273 The exocytosing lymphocytes expressed cutaneous lymphocyte antigen (CLA). 273 Apoptosis and residual filamentous bodies are present on electron microscopy. 254
Lichen planus actinicus is a distinct clinical variant of lichen planus in which lesions are limited to sun-exposed areas of the body.283. and 284. It has a predilection for certain races, 285 particularly young individuals of Oriental origin. There is some variability in the clinical expression of the disease in different countries and this has contributed to the proliferation of terms used – lichen planus tropicus, 286 lichen planus subtropicus, 287 lichenoid melanodermatitis, 288 and summertime actinic lichenoid eruption (SALE).284. and 289. More recently it has been suggested that SALE is an actinic variant of lichen nitidus. The development of pigmentation in some cases290 has also led to the suggestion that there is overlap with erythema dyschromicum perstans (see above). 265 The pigmentation may take the form of melasma-like lesions.291. and 292. Such lesions have also been reported in childhood cases. 74 Lesions have been induced by repeated exposure to ultraviolet radiation. 293
A rare erythematous variant has been described in a patient with chronic active hepatitis B infection. 294
Various treatments have been used including hydroxychloroquine, intralesional corticosteroids combined with topical sunscreens, and retinoids. Oral cyclosporine has also been used. 295
The appearances resemble lichen planus quite closely, 296 although there is usually more marked melanin incontinence284. and 293. and there may be focal parakeratosis. 283 The inflammatory cell infiltrate in lichen planus actinicus is not always as heavy as it is in typical lesions of lichen planus.
Numerous immunoglobulin-coated cytoid bodies are usually present on direct immunofluorescence. 297
Lichen planopilaris (follicular lichen planus) is a clinically heterogeneous variant of lichen planus in which keratotic follicular lesions are present, often in association with other manifestations of lichen planus.298.299. and 300. It typically affects middle-aged women and men. The annual incidence in four US hair research centers varied from 1.15% to 7.59% of new cases, reflecting its relative rarity. 301 The most common and important clinical group is characterized by scarring alopecia of the scalp, which is generalized in about half of these cases. The keratotic follicular lesions and associated erythema are best seen at the margins of the scarring alopecia. 302 In this group, changes of lichen planus are present or develop subsequently in approximately 50% of cases. 302 Rare cases have been reported in children. 303
The Graham Little–Piccardi–Lassueur syndrome is a rare but closely related entity in which there is cicatricial alopecia of the scalp, follicular keratotic lesions of glabrous skin, and variable alopecia of the axillae and groins.300.304.305. and 306. It has been reported in a patient with androgen insensitivity syndrome (testicular feminization). 307
Two other clinical groups occur but they have not received as much attention. 299 In one, there are follicular papules, without scarring, usually on the trunk and extremities. In the other, which is quite rare, there are plaques with follicular papules, usually in the retroauricular region, although other sites can be involved. 299 This variant has been called lichen planus follicularis tumidus. 308
Rare variants of lichen planopilaris include a linear form309.310.311. and 312. and the presence of lesions confined to the vulva. 313 It has been reported in a patient with erythema dyschromicum perstans, 314 and in another with scleroderma en coup de sabre. 315 It has developed in a patient receiving etanercept therapy. 316
Topical corticosteroid therapy (usually high-potency form) and intralesional steroids are the treatments of choice for patients with localized disease, particularly in the early phase.317. and 318. In one series of 30 cases treated with topical corticosteroids, resolution of the inflammatory process and blocking of the cicatricial progression were observed in 66% of cases. 317 A mild reduction occurred in 20% of patients, and no response in 13%. 317 Oral hydroxychloroquine is often used. 319 Tetracyclines appear to be more effective than once thought. 318 Cyclosporine was effective in a patient with the Graham Little–Piccardi–Lassueur syndrome. 306 It has also been used in other forms of lichen planopilaris unresponsive to corticosteroids and hydroxychloroquine. 319 Hair transplants and scalp reductions may be used in inactive end-stage disease. 318
In lichen planopilaris, there is a lichenoid reaction pattern involving the basal layer of the follicular epithelium, with an associated dense perifollicular infiltrate of lymphocytes and a few macrophages (Fig. 3.9). The changes involve the infundibulum and the isthmus of the follicle. A recent study has pointed out that this is the so-called bulge region of the follicle where the stem cells reside. 320 It is the prototypical lymphocytic cicatricial alopecia. 301 Unlike lupus erythematosus, the infiltrate does not extend around blood vessels of the mid and deep plexus. There is also some mucin in the perifollicular fibroplasia, unlike lupus erythematosus in which it is predominantly in the interfollicular dermis. 321 The interfollicular epidermis is involved in up to one-third of cases with scalp involvement,299.302. and 322. and in the rare plaque type (see above). It is not usually involved in the variant with follicular papules on the trunk and extremities. 299 If scarring alopecia develops there is variable perifollicular fibrosis and loss of hair follicles which are replaced by linear tracts of fibrosis. 322 There is also loss of the arrector pili muscles and sebaceous glands. 321 The papillary dermis may also be fibrosed. In advanced cases of scarring alopecia, the diagnostic features may no longer be present. The term ‘pseudopelade’ has been used by some for cases of end-stage scarring alopecia.
Lichen planopilaris. The lichenoid infiltrate is confined to a perifollicular location. Two different cases – (A) and (B). (H & E)
Direct immunofluorescence shows colloid bodies containing IgG and IgM in the dermis adjacent to the upper portion of the involved follicles.300. and 323. In one report, linear deposits of immunoglobulins were found along the basement membrane of the hair follicles (of the scalp) in all cases. Fibrin was present in one case; cytoid bodies were not demonstrated. 324 It should be noted that the lesions were of long standing (3–7 years). 324
This rare disease is characterized by the coexistence of lichen planus and a heterogeneous group of subepidermal blistering diseases resembling bullous pemphigoid.325.326. and 327. There are tense bullae, often on the extremities, which may develop in normal or erythematous skin or in the lesions of lichen planus.328.329.330. and 331. They do not necessarily recur with subsequent exacerbations of the lichen planus.332. and 333. Oral lesions are exceedingly rare. 334 Lichen planus pemphigoides has been reported in children.332.333. and 335. Rare clinical presentations include a unilateral distribution and onset following PUVA therapy.336. and 337. Similar lesions have been induced by the anti-motion sickness drug cinnarizine and by the ACE inhibitor ramipril.338.339. and 340. Some cases have been reported in association with neoplasia, sharing this characteristic with paraneoplastic pemphigus. 341
Lichen planus pemphigoides is different from bullous lichen planus103. and 342. in which vesicles or bullae develop only in the lichenoid papules, probably as a result of unusually severe basal damage and accompanying dermal edema.343. and 344.
The pathogenesis of lichen planus pemphigoides appears to be due to epitope spreading. It has been suggested that damage to the basal layer in lichen planus may expose or release a basement membrane zone antigen, which leads to the formation of circulating antibodies and consequent blister formation.345.346. and 347. The target antigen is a novel epitope (MCW-4) within the C-terminal NC16A domain of the 180 kDa bullous pemphigoid antigen (BP180, type XVII collagen).348.349. and 350.
Lichenoid erythrodermic bullous pemphigoid is a rare disease reported in African patients. It differs from lichen planus pemphigoides by the presence of a desquamative erythroderma and frequent mucosal lesions. 351
Lichen planus pemphigoides may be treated with topical corticosteroids, systemic steroids, tetracycline and nicotinamide combined, retinoids, and dapsone. 352 Systemic corticosteroids appear to be the most effective treatment for extensive disease. 352
A typical lesion of lichen planus pemphigoides consists of a subepidermal bulla which is cell poor, with only a mild, perivascular infiltrate of lymphocytes, neutrophils, and eosinophils. 353 The presence of neutrophils and eosinophils has not been mentioned in all reports. Sometimes a lichenoid infiltrate is present at the margins of the blister343 and there are occasional degenerate keratinocytes in the epidermis overlying the blister. 328 Lesions which arise in papules of lichen planus show predominantly the features of lichen planus; a few eosinophils and neutrophils are usually present, in contrast to bullous lichen planus in which they are absent. 328 In one report, a pemphigus vulgaris-like pattern was present in the bullous areas. 354
Direct immunofluorescence of the bullae will usually show IgG, C3, and C9 neoantigen in the basement membrane zone and there is often a circulating antibody to the basement membrane zone.355. and 356. Indirect split-skin immunofluorescence has shown binding to the roof of the split. 346
In lichen planus pemphigoides the split occurs in the lamina lucida, as it does in bullous pemphigoid.346. and 357. Immunoelectron microscopy has shown that the localization of the immune deposits may resemble that seen in bullous pemphigoid, cicatricial pemphigoid, or epidermolysis bullosa acquisita, evidence of a heterogeneous disorder. 358
Keratosis lichenoides chronica is characterized by violaceous, papular, and nodular lesions in a linear and reticulate pattern on the extremities and a seborrheic dermatitis-like facial eruption.359.360.361.362.363.364. and 365. A rare vascular variant with telangiectasias has been reported. 366 Oral ulceration and nail involvement may occur.367. and 368.
It is a rare condition, particularly in children.369.370.371. and 372. It has been suggested that pediatric-onset disease is different from adult-onset keratosis lichenoides chronica. 373 Childhood cases may have familial occurrence, and probably autosomal recessive inheritance. Early or congenital onset with facial erythemato-purpuric macules is sometimes seen. 373 Forehead, eyebrow, and eyelash alopecia are usually present. A case mimicking verrucous secondary syphilis has been reported.374. and 375. The condition is possibly an unusual chronic variant of lichen planus, although this concept has been challenged.376. and 377. Böer believes that there is an authentic and distinctive condition that should continue to be called keratosis lichenoides chronica, but that many of the purported cases are lichen planus, lupus erythematosus, or lichen simplex chronicus. 378
Keratosis lichenoides chronica may be associated with internal diseases such as glomerulonephritis, hypothyroidism, and lymphoproliferative disorders.366.376. and 379. In one patient with multiple myeloma there were eruptive keratoacanthoma-like lesions. 380
The disease is refractory to many different treatment modalities, 366 although calcipotriol or tacalcitol alone, or in combination with oral retinoids, may give good results. 366 Oral retinoids alone are sometimes effective. 381 Phototherapy has also been used. 382
There is a lichenoid reaction pattern with prominent basal cell death and focal basal vacuolar change. 383 The inflammatory infiltrate usually includes a few plasma cells and sometimes there is deeper perivascular and periappendageal cuffing. 384 Telangiectasia of superficial dermal vessels is sometimes noted. 385 Epidermal changes are variable, with alternating areas of atrophy and acanthosis sometimes present, as well as focal parakeratosis. 386 The parakeratosis often has a staggered appearance with neutrophil remnants. 378 Cornoid lamellae and amyloid deposits in the papillary dermis have been recorded. 377 Numerous IgM-containing colloid bodies are usually found on direct immunofluorescence. 367
The term ‘lichen planoporitis’ was used for a case with the clinical features of keratosis lichenoides chronica and histological changes that included a lichenoid reaction centered on the acrosyringium and upper eccrine duct with focal squamous metaplasia of the upper duct and overlying hypergranulosis and keratin plugs. 387 Ruben and LeBoit have also reported eccrine duct involvement in a case of keratosis lichenoides chronica. 388 Böer states that the lichenoid infiltrate in keratosis lichenoides chronica is commonly centered around infundibula and acrosyringia. 378
Lupus erythematosus–lichen planus overlap syndrome is a heterogeneous entity in which one or more of the clinical, histological, and immunopathological features of both diseases are present.389. and 390. Some cases may represent the coexistence of lichen planus and lupus erythematosus, while in others the ultimate diagnosis may depend on the course of the disease.391. and 392. In most cases, the lupus erythematosus is of the chronic discoid or systemic type; rarely, it is of the subacute type. 393 It was the cause of a scarring alopecia in one case. 394 Before the diagnosis of an overlap syndrome is entertained, it should be remembered that some lesions of cutaneous lupus erythematosus may have numerous Civatte bodies and a rather superficial inflammatory cell infiltrate which at first glance may be mistaken for lichen planus. The use of an immunofluorescent technique using patient’s serum and autologous lesional skin as a substrate may assist in the future in elucidating the correct diagnosis in some of these cases. 395
Lichen nitidus is a rare, usually asymptomatic chronic eruption characterized by the presence of multiple, small flesh-colored papules, 1–2 mm in diameter.396. and 397. The lesions have a predilection for the upper extremities, chest, abdomen, and genitalia of children and young adult males.396. and 398. The disorder is most often localized but sometimes lesions are more generalized.399.400. and 401. Familial cases are rare. 402 It has been reported in association with Down syndrome. 403 Nail changes404. and 405. and involvement of the palms and soles406.407. and 408. have been reported. It has been suggested that cases reported in the past as summertime actinic lichenoid eruption (SALE) should be reclassified as actinic lichen nitidus.409.410. and 411. It has been reported in association with lichen spinulosus. 412
Although regarded originally as a variant of lichen planus, lichen nitidus is now considered a distinct entity of unknown etiology. It has followed hepatitis B vaccine. 413 The lymphocytes in the dermal infiltrate in lichen nitidus express different markers from those in lichen planus. 414 Lichen planus has developed subsequent to generalized lichen nitidus in a child. 415
Although spontaneous remissions of lichen nitidus are common, 399 persistent lesions and those that are refractory to various treatments can pose therapeutic challenges. Sometimes resolution is accompanied by postinflammatory hyperpigmentation. 399 Some of the treatments used include systemic and topical corticosteroids, antihistamines, retinoids, low-dose cyclosporine, itraconazole, isoniazid, and ultraviolet therapy.400. and 416. Generalized lichen nitidus has been successfully treated with narrowband-UVB phototherapy.400. and 417.
A papule of lichen nitidus shows a dense, well-circumscribed, subepidermal infiltrate, sharply limited to one or two adjacent dermal papillae. 396 Claw-like, acanthotic rete ridges, which appear to grasp the infiltrate, are present at the periphery of the papule (Fig. 3.10). The inflammatory cells push against the undersurface of the epidermis, which may be thinned and show overlying parakeratosis. Occasional Civatte bodies are present in the basal layer.
Lichen nitidus. (A) There are two discrete foci of inflammation involving the superficial dermis. (B) Claw-like downgrowths of the rete ridges are present at the margins of these foci. (C) Another case with a broader lesion. (H & E)
In addition to lymphocytes, histiocytes, and melanophages, there are also epithelioid cells and occasional multinucleate giant cells in the inflammatory infiltrate. 418 Rarely, plasma cells are conspicuous. 419 The appearances are sometimes frankly granulomatous and these lesions must be distinguished from disseminated granuloma annulare in which the infiltrate may be superficial and the necrobiosis sometimes quite subtle. Lichen nitidus also needs to be distinguished from an early lesion of lichen scrofulosorum. While the infiltrate in lichen nitidus ‘hugs’ the epidermis and expands the dermal papilla, the granulomas in lichen scrofulosorum do not cause widening of the papillae. Furthermore, in lichen scrofulosorum there may be mild spongiosis and exocytosis of neutrophils into the epidermis. 420
Rare changes that have been reported include subepidermal vesiculation, 421 transepidermal elimination of the inflammatory infiltrate,418.422. and 423. and the presence of perifollicular granulomas. 424 Periappendageal inflammation mimicking lichen striatus has also been reported. 425
Direct immunofluorescence is usually negative, a distinguishing feature from lichen planus.
The ultrastructural changes in lichen nitidus are similar to those of lichen planus. 426
Lichen striatus is a linear, papular eruption of unknown etiology which may extend in a continuous or interrupted fashion along one side of the body, usually the length of an extremity.427. and 428. Annular429 and bilateral forms430 have been reported. The lesions often follow Blaschko’s lines;431.432.433. and 434. this is almost invariable for lesions on the trunk and face. 435 Nail changes are not uncommon,436. and 437. resulting in onychodystrophy. 438 Lichen striatus has a predilection for female children and adolescents. 435 Familial cases are rare. 439 An unusual presentation in children is the presence of linear lesions on the nose with some overlap features with lupus erythematosus. 440 Spontaneous resolution usually occurs after 6 months, although some cases persist longer. 435 Hypochromic sequelae occur in nearly 30% of cases; hyperchromic sequelae are much less common. 435 Relapses are uncommon. A history of atopy is sometimes present in affected individuals.435. and 441.
Lichen striatus has been reported following BCG vaccination, 434 varicella infection, 442 solarium exposure, 443 and following a flu-like illness. 444 It has also developed in a pregnant woman, 445 and in a patient with plaque psoriasis. 446 It has been suggested that lichen striatus represents an autoimmune CD8-mediated response against a mutate keratinocytic clone, which represents a somatic mutation occurring after fertilization. 435
Because lichen striatus usually resolves spontaneously, therapy is not required. 160 Topical steroids have been used to treat the disease, but they do not appear to influence the duration of the lesions. 435 Tacrolimus (0.1%) is an effective treatment option for lichen striatus of the face and other areas.447.448. and 449. Pimecrolimus cream has also been effective in adult patients.450.451. and 452.
There is a lichenoid reaction pattern with an infiltrate of lymphocytes, histiocytes, and melanophages occupying three or four adjacent dermal papillae. 455 The overlying epidermis is acanthotic with mild spongiosis associated with exocytosis of inflammatory cells. Small intraepidermal vesicles containing Langerhans cells are present in half of the cases. 456 Dyskeratotic cells are often present at all levels of the epidermis; such cells are uncommon in linear lichen planus. 457 There is usually mild hyperkeratosis and focal parakeratosis. 427 The dermal papillae are mildly edematous. The infiltrate is usually less dense than in lichen planus and it may extend around hair follicles or vessels in the mid-plexus. Eccrine extension of the infiltrate is often present (Fig. 3.11).453. and 454. A monoclonal population of T cells has been reported in one case but excluded in others. 458
Lichen striatus. This case has a florid perieccrine infiltrate of lymphocytes. (H & E)
It should not be forgotten that lichen striatus has been called a ‘chameleon’. 456 The histology may closely mimic lichen nitidus or lichen planus even though the clinical features are those of lichen striatus. Furthermore, adult blaschkitis has histological overlap features with lichen striatus. 459
Dyskeratotic cells similar to the corps ronds of Darier’s disease have been described in the upper epidermis. 454 The Civatte bodies in the basal layer show the usual changes of apoptosis on electron microscopy.
Lichen planus-like keratosis (LPLK) is a commonly encountered entity in routine histopathology.460.461.462.463.464.465. and 466. Synonyms used for this entity include solitary lichen planus, benign lichenoid keratosis, 461 lichenoid benign keratosis,462. and 463. and involuting lichenoid plaque. 464 It should not be confused with lichenoid actinic (solar) keratosis467 in which epithelial atypia is a prerequisite for diagnosis. 468 Lichen planus-like keratoses are usually solitary, discrete, slightly raised lesions of short duration, measuring 3–10 mm in diameter. Multiple lesions (20–40) have been reported, but the illustration of one such case appears to show a cornoid lamella. 469 In a study of 1040 cases, 8% of patients presented with two lesions, and less than 1% with three lesions. 470 The sudden appearance of the lesion is often a striking feature. Lesions are violaceous or pink, often with a rusty tinge. 460 There may be a thin, overlying scale. The dermoscopic features correlate with the stage of the lesion and the nature of the lesion being regressed.471. and 472. There is a predilection for the arms and presternal area of middle-aged and elderly women. 470 Lesions are sometimes mildly pruritic or ‘burning’. 461 Clinically, LPLK is usually misdiagnosed as a basal cell carcinoma or Bowen’s disease.
Lichen planus-like keratosis is a heterogeneous condition which usually represents the attempted cell-mediated immune rejection of any of several different types of epidermal lesion. In most instances this is a solar lentigo,460.473. and 474. but in some lesions there is a suggestion of an underlying seborrheic keratosis, large cell acanthoma or even a viral wart. 475 Constant pressure was incriminated in one case. 476 Acetaminophen (paracetamol) was incriminated in another case. 477 In one study, a contiguous solar lentigo was present in only 7% of cases and a seborrheic keratosis in 8.4%. 465 These findings do not accord with the author’s own experiences.
There is a florid lichenoid reaction pattern with numerous apoptotic keratinocytes in the basal layer and accompanying mild vacuolar change (Fig. 3.12). The infiltrate is usually quite dense and often includes a few plasma cells, eosinophils, and even neutrophils in addition to the lymphocytes and macrophages. The infiltrate may obscure the dermoepidermal interface. A rare variant of LPLK has histological features simulating mycosis fungoides. 478 Pautrier-like microabscesses, the alignment of lymphocytes along the basal layer, and epidermotropism are features of this variant. 478 Some of the cells are CD30+. 470
(A) Lichen planus-like keratosis. (B) There is a lichenoid reaction pattern with some deeper extension of the infiltrate than is usual in lichen planus. (H & E)
Pigment incontinence may be prominent. This is so in the late (regressed, atrophic) stage; epidermal atrophy and papillary dermal fibrosis are also present. 470 There may be mild atypia of keratinocytes but this is never as marked as it is in a lichenoid solar keratosis. 32 Before a diagnosis of LPLK is made, the sections should be carefully scanned to ensure that there is not an underlying melanocytic proliferation. 479
There is often mild hyperkeratosis and focal parakeratosis. 465 The presence of parakeratosis allows a distinction to be made with lichen planus. Hypergranulosis is not as pronounced as in lichen planus. A contiguous solar lentigo or large cell acanthoma (Fig. 3.13) is sometimes seen. 480
Lichen planus-like keratosis. It is arising in a large cell acanthoma. (H & E)
Usually cell death is scattered and of apoptotic type. At times confluent necrosis occurs and in these cases subepidermal clefting may result. Sometimes this variant simulates toxic epidermal necrolysis (Fig. 3.14). At other times, the lesions are bullous with a heavy lymphocytic infiltrate and increased numbers of dead basal keratinocytes. 470There is also a rare ‘creeping’ form in which there is focal acute activity and other areas with few lymphocytes, little if any apoptotic cell death, and some melanin incontinence. A careful search for a cornoid lamella of porokeratosis should always be made when these changes are present.
Lichen planus-like keratosis of the toxic epidermal necrolysis type. (A) There is blister formation in this region. (B) Another area of the same case. (H & E)
The attempt to classify ‘lichenoid keratoses’ into three groups – lichen planus-like keratosis, seborrheic keratosis-like lichenoid keratosis, and lupus erythematosus-like lichenoid keratosis – deserves some comment. 481 The seborrheic keratosis-like variant is best called an irritated (lichenoid) seborrheic keratosis and the lupus erythematosus-like variant is simply a lesion with some basal clefting (see above). It may sometimes represent early lupus erythematosus. 482 Notwithstanding this comment, the author admits that making a distinction between LPLK and lupus erythematosus is occasionally difficult for lesions on the face in which the biopsy is small. The presence of follicular involvement in lupus erythematosus and its absence in LPLK is not a reliable point of distinction as some LPLKs can have not only involvement of the infundibula of follicles, but also follicular involvement at a slightly deeper level.
A summary of the histological types of LPLK is shown in Table 3.4.
Direct immunofluorescence shows colloid bodies containing IgM and some basement membrane fibrin. 462 Immunohistochemistry shows fewer Langerhans cells in the epidermis than in lichen planus. 483 The infiltrate is polyclonal. 484
Mention should be made of the report of Melan A-positive pseudonests in the setting of lichenoid inflammation. 485 These nests did not stain for S100 protein or a ‘melanoma cocktail’. 485 A subsequent paper found no Melan A/MART-1 positive pseudonests in lichenoid inflammation. 486
Shave excision is the usual method of treatment for these lesions.
A lichenoid eruption has been reported following the ingestion of a wide range of chemical substances and drugs.487. and 488. The eruption may closely mimic lichen planus clinically, although at other times there is eczematization and more pronounced, residual hyperpigmentation. Rarely the eruption follows Blaschko’s lines. 489 Some of the β-adrenergic blocking agents produce a psoriasiform pattern clinically but lichenoid features histologically.490. and 491. Discontinuation of the drug usually leads to clearing of the rash over a period of several weeks. 492 A lichenoid reaction has developed in a temporary henna tattoo, 493 as well as in permanent tattoos.494. and 495.
Lichenoid eruptions have been produced by gold, 496 gold-containing liquor,497. and 498. methyldopa, 499β-adrenergic blocking agents, 500 penicillamine, 501 quinine, 502 quinidine, 503 synthetic antimalarials, 504 and ethambutol. 505 Less common causes of a lichenoid reaction are captopril, 506 enalapril,507. and 508. amlodipine, 509 naproxen,510.511. and 512. dapsone, simvastatin, 513 pravastatin, 514 indapamide, arsenicals, mercury, 515 iodides, imatinib,516. and 517. etanercept, 518 adalimumab, 519 infliximab, 520 acetylsalicylic acid, 521 orlistat, 522 ticlopidine, 523 sodium valproate, 524 nicorandil, 525 terazosin, 526 carbamazepine, 527 phenothiazine derivatives, salsalate, 528 chlorpropamide, 529 suramin, 530 tiopronin, 531 docetaxel, 532 dactinomycin, 533 doxorubicin,534.535.536. and 537. cyanamide, 538 omeprazole, lansoprazole, pantoprazole, 539 spironolactone, 540 metformin, 541 glimepiride, 542 indoramin, 543 interferon alfa-2b, intravenous immunoglobulin, 544 and streptomycin. 545Photodistributed lesions may occur with thiazides, 546 some tetracyclines, 547 sparfloxacin, 548 nimesulide, 549 torsemide (a loop diuretic), 550 clopidogrel, 551 solifenacin, 552 capecitabine, 553 diltiazem,554.555. and 556. ethambutol, 160 quinine, 160 pyritinol, 160 carbamazepine, 160 and pyrazinamide. 557 Hyperpigmentation may follow. 556 A lichenoid stomatitis, which may take time to clear after cessation of the drug, can be produced by methyldopa and rarely by lithium carbonate558 or propranolol. Contact with color film developer may produce a lichenoid photodermatitis. 492 A topical mixture of the local anesthetic agents lidocaine and prilocaine produced basal clefting similar to epidermolysis bullosa simplex with basophilic granules in the cleft.559. and 560. A lichen planus-like eruption has been reported at sites repeatedly exposed to methacrylic acid esters used in the car industry561 and also at injection sites of granulocyte colony-stimulating factor (GCSF). 562 A severe lichenoid eruption and stomatitis developed in a patient receiving GCSF, interferon-α-2A, and ribavirin. 563 Voriconazole has induced a blistering eruption with histological lichenoid features. The eruption occurred in the setting of graft-versus-host disease. 564 A list of the drugs that produce lichenoid eruptions is included in Table 3.5.
Lichenoid drug eruptions usually differ from lichen planus by the presence of focal parakeratosis and mild basal vacuolar change, as well as a few eosinophils and sometimes plasma cells in the infiltrate (Fig. 3.15). These features are said to be more common in non-photodistributed lesions, whereas photodistributed lesions may mimic lichen planus. 566 Apoptotic keratinocytes may be found above the basal layer in lichenoid drug reactions, an uncommon feature in lichen planus. 567 There is often more melanin incontinence than in lichen planus. The infiltrate is often less dense and less band-like than in lichen planus itself. A few inflammatory cells may extend around vessels in the mid and lower dermis. Sometimes the histological features closely simulate those of lichen planus. 566 A few eosinophils in the infiltrate may be the only clue to the diagnosis.
Lichenoid drug eruption. There is focal parakeratosis and a few eosinophils in the infiltrate. Early changes of photosensitivity are apparent. (H & E)
An unusual lichenoid reaction with epidermotropic multinucleate giant cells in the inflammatory infiltrate has been reported in patients taking a variety of drugs. 568 The term ‘giant cell lichenoid dermatitis’ has been used for this pattern (Fig. 3.16). 569 One patient subsequently developed sarcoidosis. 569‘Lichenoid sarcoidosis’ has been used for a case with both lichenoid and sarcoidal features. 570 Giant cell lichenoid dermatitis has recently been reported in herpes zoster scars, particularly in bone marrow recipients. 571 A giant cell lichenoid dermatitis was present in a patient with baboon syndrome (see p. 117). It followed the intravenous administration of amoxicillin-clavulanic acid. 572
Giant cell lichenoid dermatitis. There are several multinucleate giant cells in the lichenoid infiltrate. (H & E)
A fixed drug eruption is a round or oval erythematous lesion which develops within hours of taking the offending drug and which recurs at the same site with subsequent exposure to the drug. 573 Lesions may be solitary or multiple. A bullous variant with widespread lesions also occurs.574.575. and 576. Fixed eruptions subside on withdrawal of the drug, leaving a hyperpigmented macule. 577 Non-pigmenting lesions have been described578.579. and 580. and depigmented areas may be the result in patients whose skin is naturally heavily pigmented. Sometimes there is a burning sensation in the erythematous lesions, but systemic manifestations such as malaise and fever are uncommon. 573 Rare clinical variants include eczematous, necrotizing, 581 cellulitis-like, 582‘Dalmatian dog’-like, 583 urticarial, linear,584. and 585. ‘wandering’, 586 butterfly rash-like, 587 and erythema dyschromicum perstans-like588 types. A unilateral eruption of the breast has been reported. 589 In two cases the eruption occurred at the sites of ear piercing. 590 An interesting presentation concerns the fixed drug eruption that developed in a male after coitus, which was thought to have resulted from the trimethoprim-sulfamethoxazole that his wife was taking. 591 Other ‘sexually transmitted’ cases have since been reported. 592 Common sites of involvement include the face, lips, 593 buttocks, and genitals.594. and 595. Paronychia is a rare presentation. 596
In one series of 446 cases of drug eruption, 92 (21%) were instances of fixed drug eruptions and, of these, 16 were bullous and generalized. 597 Over 100 drugs have been incriminated, but the major offenders are sulfonamides, 598 particularly the combination of trimethoprim-sulfamethoxazole,591.599. and 600. tetracyclines,600.601. and 602. tranquilizers, quinine, phenolphthalein (in laxatives), and some analgesics.603. and 604. In some countries, antituberculous drugs and antimalarials are a major cause of a fixed drug eruption. 605 Specific drugs incriminated in the literature include minocycline, 602 nystatin, 606 colchicine, 607 clioquinol, 608 penicillin, 609 amoxicillin, 610 erythromycin, 611 clarithromycin, 612 ciprofloxacin, 613 vancomycin, 614 rifampicin, 604 tranexamic acid, 615 griseofulvin, 616 terbinafine, 617 dimenhydrinate (Gravol),618.619. and 620. diphenhydramine, 621 levocetirizine, 622 paclitaxel, 623 topotecan, 582 temazepam, 624 mefenamic acid,575.583. and 625. carbamazepine,626. and 627. acetylsalicylic acid, 628 acetaminophen (paracetamol),628.629.630.631.632.633.634.635. and 636. fluconazole,637. and 638. ketoconazole, 639 itraconazole, 639 feprazone, 640 tartrazine, 641 ticlopidine, 642 phenytoin, 627 lamotrigine, 643 pseudoephedrine,644. and 645. dextromethorphan, 646 iomeprol, 647 phenylpropanolamine hydrochloride, 648 tropisetron, 649 metramizole, 600 phenylbutazone, 600 etodolac, 650 metamizole, 651 naproxen,589.652. and 653. piroxicam,590. and 654. tenoxicam, 590 interferon-β-1b, 655 ibuprofen, 656 acetaminophen/indomethacin/granisetron/IL-2 in combination, 657 celecoxib, 604 chlormezanone, 604 omeprazole, 628 diltiazem, 628 enalapril, 628 lansoprazole, 628 fluoxetine, 628 amplodipine, 628 S-carboxymethyl-L-cysteine, 658 eperisone hydrochloride, 659 lactose,660. and 661. metronidazole,662.663. and 664. tinidazole, 662 nimesulide, 665 cetirizine,666. and 667. and theophylline. 588 More complete lists are included in several reviews of the subject.573.603. and 604. The drugs responsible are listed in Table 3.5. Two different drugs have been involved in the same patient.668. and 669. In yet another report, three different drugs each produced a fixed drug eruption at a different site. 670 It has been suggested that the distribution of lesions is influenced by the drug in question; tetracyclines and cotrimoxazole tend to involve the glans penis, while pyrazolones and naproxen affect mainly the lips and mucosae.599. and 671. Antimalarials mainly involve the face and lips. 605 There are rare reports of fixed food eruptions. Lentils672 and strawberries673 have been implicated. Foods may sometimes produce a flare in a fixed drug eruption. 674 The Japanese herbal drug ‘kakkon-to’ has produced an extensive fixed drug eruption. 675 A solitary lesion has been produced by the Chinese herbal medicine, ma huang (Ephedra Hebra), mainly containing pseudoephedrine and ephedrine. 676 Influenza vaccination has resulted in a generalized bullous eruption. 576 In some instances no agent can be incriminated. Such cases have been called ‘fixed drug-like eruption’. 677
Numerous studies have attempted to elucidate the pathogenesis of fixed eruptions. It appears that the offending drug acts as a hapten and binds to a protein in basal keratinocytes and sometimes melanocytes. 573 As a consequence, an immunological reaction is stimulated, which probably takes the form of an antibody-dependent, cellular cytotoxic response. 678 CD8+ T lymphocytes attack the drug-altered epidermal cells producing apoptosis; it appears to be mediated by Fas ligand (FasL), in the presence of interferon-γ, triggering a caspase cascade. 145 Drug-specific CD8+ memory T cells also play a role in preserving the cutaneous memory function which characterizes a fixed eruption.590. and 679. It appears that these memory T cells transiently acquire a natural killer-like phenotype and express cytotoxic granules upon activation. 680 Keratinocytes at the site of a fixed drug eruption express one of the cell adhesion molecules (ICAM-1) that is involved in the adherence reaction between lymphocytes and epidermal keratinocytes. 681 It has been suggested that localized expression of this adhesion antigen may be one factor that explains the site specificity of fixed drug eruptions. 681 The occurrence of some fixed drug eruptions at sites of trauma or previous inflammation may be a manifestation of an isotopic response. 590 It is also possible that preformed cytokines may exist at such sites. These findings have been incorporated into the following hypothesis: that the causative drug activates mast cells or keratinocytes to release cytokines or induces keratinocytes to express adhesion molecules on their surfaces, leading to the activation of epidermal CD8+ T cells, 682 and resulting in death of keratinocytes by a FasL-mediated pathway. 145 This theory has been put forward in an attempt to explain the early onset of symptoms, which occur much earlier than traditional cell-mediated responses could produce.
Although CD8+ cells are more numerous than CD4+ cells in the epidermis and dermis, cells which are CD25+ CD4+ appear to migrate into the epidermis of active lesions and exert a regulatory function by releasing interleukin-10, resulting in the resolution of the lesion(s). 145 These same cells have also been shown to be involved in the induction of desensitization to a fixed drug eruption. 145
There appears to be a genetic susceptibility to fixed drug eruptions, with an increased incidence of HLA-B22683 and HLA-A30 B13 Cw6. 684
Established lesions show a lichenoid reaction pattern with prominent vacuolar change and Civatte body formation (Fig. 3.17). The degenerate keratinocytes usually show less shrinkage than in lichen planus. The inflammatory infiltrate tends to obscure the dermoepidermal interface, as in erythema multiforme and some cases of pityriasis lichenoides et varioliformis acuta (PLEVA). The infiltrate often extends into the mid and upper epidermis producing death of keratinocytes above the basal layer (Fig. 3.18). Fixed drug eruptions can usually be distinguished from erythema multiforme by the deeper extension of the infiltrate, the presence of a few neutrophils, and the more prominent melanin incontinence in fixed eruptions.
Fixed drug eruption. Dead keratinocytes are present in the basal layer and at higher levels of the epidermis. Lymphocytes extend into the epidermis. (H & E)
Fixed drug eruption. The infiltrate of lymphocytes extends some distance into the epidermis, resulting in cell death in the basal layer and above. (H & E)
Based on one study, 685 it appears that very early lesions may show epidermal spongiosis, dermal edema and neutrophil microabscesses and numerous eosinophils in the dermis. These features have usually disappeared after several days, although some eosinophils persist.
The clinical variants of fixed drug eruption have not been documented very well, except for the bullous form which results when subepidermal clefting occurs. This may be misdiagnosed as erythema multiforme. Spongiotic vesiculation is present in the eczematous variant and a picture resembling an urticarial reaction is seen in others. Vasculitis is another pattern seen, rarely, in fixed drug eruptions. 632 In the non-pigmenting variant there is a mild perivascular and interstitial mixed inflammatory infiltrate in the dermis. 579
There is prominent clumping of tonofilaments in the cytoplasm of basal keratinocytes, which provides an explanation for the bright eosinophilic cytoplasm and the comparatively small amount of shrinkage that these cells undergo during cell death. 577 There is also condensation of nuclear chromatin. Intracytoplasmic desmosomes are sometimes seen. Filamentous bodies, composed of filaments which are less electron dense than in the intact cells, are quite numerous. 686 These contain some melanosomes and sometimes nuclear remnants. 577 They are sometimes phagocytosed by adjacent keratinocytes or macrophages. 577 The accumulation of tonofilaments may represent a response by keratinocytes to sublethal injury or some other stimulus. It has the effect of masking the exact mode of death – apoptosis or necrosis. The term ‘filamentous degeneration’ has some merit in these circumstances.
Erythema multiforme is a self-limited, sometimes episodic disease of the skin, which may also involve the mucous membranes. It is characterized by a pleomorphic eruption consisting of erythematous macules, papules, urticarial plaques, vesicles, and bullae.687. and 688. Individual lesions may evolve through a papular, vesicular and target (iris) stage in which bullae surmount an erythematous maculopapule. 689 Lesions tend to be distributed symmetrically with a predilection for the extremities, particularly the hands.
In the past, erythema multiforme was classified into erythema multiforme minor and erythema multiforme major, the latter being characterized by a severe and sometimes fatal illness in which fever, systemic symptoms and severe oral lesions were usually present.689.690.691. and 692. The term Stevens–Johnson syndrome was also applied to these severe cases with oral involvement. Recently, an attempt has been made to distinguish Stevens–Johnson syndrome from erythema multiforme major with mucosal lesions on the basis of their different cutaneous lesions and their etiology; 693 their mucosal lesions are similar. Stevens–Johnson syndrome is said to be characterized by flat atypical target lesions or purpuric macules that are widespread or limited to the trunk. Erythema multiforme major with mucosal lesions has typical or raised atypical target lesions, located on the extremities and/or the face. 693 Using these definitions, Stevens–Johnson syndrome is usually related to drugs and erythema multiforme to herpes or other infections.693. and 694.
In a prospective European study involving 552 patients, erythema multiforme major was found to differ from Stevens–Johnson syndrome and toxic epidermal necrolysis not only in severity but also in several demographic features. 695 Erythema multiforme major occurred in younger males, had frequent recurrences, less fever, milder mucosal lesions, and a lack of association with collagen vascular diseases, HIV infection, or cancer. 695 Recent or recurrent herpes simplex infection was the principal risk factor. 695
The criteria used to distinguish the component diseases that form this spectrum have been criticized on the basis that they ignore the fundamental clinical differences between these two related conditions. 696 Namely, Stevens–Johnson syndrome is associated with systemic symptoms and involvement of internal organs, whereas erythema multiforme is not. 696Toxic epidermal necrolysis (see below) has variously been regarded as a separate entity or as representing the severe end of the spectrum of erythema multiforme major or Stevens–Johnson syndrome.697. and 698. Some clinicians have arbitrarily diagnosed toxic epidermal necrolysis when blisters and peeling involved more than 30% of the total body surface area and Stevens–Johnson syndrome when mucosal lesions were present and blistering involved less than 30% of the body surface. 699 An international group has attempted to standardize the terminology by defining five clinical categories – bullous erythema multiforme, Stevens–Johnson syndrome, overlap Stevens–Johnson syndrome/toxic epidermal necrolysis, toxic epidermal necrolysis with ‘spots’ (widespread purpuric macules or target lesions), and toxic epidermal necrolysis without ‘spots’.699. and 700. The strategy behind this approach is summed up by two of the experts in this area: ‘Our current concept is to separate an EM spectrum (EM minor combined with EM major), from an SJS/TEN spectrum.’701 Many clinicians use only three categories – erythema multiforme, Stevens–Johnson syndrome, and toxic epidermal necrolysis – making comparative studies of this clinical spectrum difficult.
Two further clinical subgroups have been delineated: recurrent erythema multiforme and a rare persistent form. The persistent form has been associated with an underlying malignancy and with Epstein–Barr virus infection.702. and 703. Some cases are idiopathic. 704 The recurrent form is frequently associated with recurring infections, 705 often with the herpes simplex virus.706. and 707. Rarely it is associated with hepatitis C infection. 708 Sometimes the recurrent lesions mimic polymorphic light eruption by having a photodistribution. 709 Patients with certain HLA types – B35, B62 (B15), and DR53 – are more susceptible to this recurrent form. 710
Unusual clinical presentations include the limitation of lesions to areas of lymphatic obstruction, 711 to nevi712. and 713. or to Blaschko’s lines, 714 a photosensitive eruption, 715 and the development of eruptive nevocellular nevi following severe erythema multiforme. 716 Long-term follow-up suggests that nevi which develop after bullous disorders are more likely to remain benign compared with those in patients with ongoing immunosuppression. 717 Leukoderma is a rare complication. 718
Neonatal erythema multiforme may be difficult to diagnose and manage.719.720. and 721. Fortunately it is quite rare.
Over 100 different causal factors have been implicated, including viral and bacterial infections, drugs, and several associated neoplastic conditions. 722 Infection with herpes simplex type 1 is a common precipitating factor for minor forms,723.724. and 725. while Mycoplasma pneumoniae infection726. and 727. and drugs are often incriminated in the more severe cases.690. and 728. A study of erythema multiforme in children found that the minor forms were due to herpes simplex and the cases with Stevens–Johnson syndrome to M. pneumoniae infection.729.730.731. and 732. Drugs were rarely implicated in this age group. 729M. pneumoniae can produce a mucositis, as seen in Stevens–Johnson syndrome, but without skin lesions.733. and 734. Infection with Epstein–Barr virus has been associated with both minor and persistent erythema multiforme.702.735. and 736. Other infections to be associated with erythema multiforme include cytomegalovirus,737. and 738. Lyme disease (in its early stages),739. and 740. syphilis, 741 orf, 742 molluscum contagiosum, 743 various dermatophyte infections,744.745. and 746. sporotrichosis, 747 brucella infection, 748 Kikuchi disease, 749Gardnerella vaginalis infection, 750 HTLV-1 infection, and hepatitis B.751. and 752. It has also followed hepatitis B immunization, 753 and immunization with smallpox, anthrax, and tetanus vaccines, 754 and diphtheria-pertussis-tetanus vaccine. 755
Numerous drugs have been involved, most commonly the sulfonamides and non-steroidal anti-inflammatory drugs.756.757. and 758. They often produce a severe reaction. Corticosteroids may on occasion be a culprit drug. 759 Specific drugs incriminated include ciprofloxacin,760. and 761. ofloxacin, 762 nystatin, 763 the aminopenicillins, 764 doxycycline,765. and 766. cimetidine, suramin, 767 theophylline, 768 allopurinol, etretinate, 769 terbinafine,770.771.772.773. and 774. griseofulvin, 775 clonazepam, 776 acarbose, 777 ticlopidine, 642 sertraline,758. and 778. rofecoxib, 779 valdecoxib, 780 amifostine, 781 cyclophosphamide, 782 lamotrigine,758.783.784. and 785. bezafibrate, 786 cocaine, 787 phenytoin,788. and 789. progesterone in a low-dose oral contraceptive pill, 790 sennoside, 791 amifostine, 792 thalidomide, 793 ramipril, 794 the contrast medium iopentol, 795 fenoterol, 796 the multikinase inhibitor sorafenib, 797 pantoprazole, 758 tramadol, 758 irbesartan, 798 bupropion, 799 topical nitrogen mustard, 800 mefloquine, 801 trichloroethylene, 802 the Chinese herbal drug ‘Dong Ling Hou Tong Pian’, 803 and nevirapine.758.804. and 805. A recent paper indicated that allopurinol is now the most common cause of Stevens–Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. 806 The various drugs that have precipitated erythema multiforme/toxic epidermal necrolysis are listed in Table 3.5.
Erythema multiforme has been reported in association with an allergic contact dermatitis; plants, poison ivy, 807 woods, 808 paraphenylenediamine in a henna tattoo, 809 epoxy sealants, and diphencyprone (used in the treatment of warts) have been incriminated.810.811. and 812. It has also followed polymorphic light eruption. 813 An erythema multiforme-like reaction has been reported following the intravenous injection of vinblastine in the vicinity, 814 and at sites of radiation therapy in patients taking antiepileptic drugs. 815
Erythema multiforme appears to result from a cell-mediated immune reaction to one of the many agents listed above. In the case of herpes simplex, lymphocytes home to viral antigen-positive cells containing the herpes DNA polymerase gene (Pol).816. and 817. The term HAEM (herpes-associated erythema multiforme) is used for these cases. The virus often remains in affected cutaneous sites for up to 3 months or more following resolution of the erythema multiforme, suggesting that the skin may function as a site of viral persistence.818. and 819. Herpes simplex virus can be detected in up to three-quarters of patients with erythema multiforme, on paraffin-embedded biopsy material.820.821. and 822. Two classes of lymphocytes appear to be involved in this cell-mediated reaction. They are T lymphocytes carrying the Vβ2 phenotype823 and CD8+ cells with natural killer-cell activity. 824 CD4+ T lymphocytes appear to be more important in erythema multiforme and CD8+ cells in Stevens–Johnson syndrome/toxic epidermal necrolysis. 825 The effector cytokine is interferon-γ in cases of HAEM, while in drug-induced erythema multiforme major/Stevens–Johnson syndrome/toxic epidermal necrolysis, tumor necrosis factor (TNF-α), perforin, and granzyme B produce the epidermal destruction.757.826.827. and 828. This is a simplistic explanation of the pathogenesis as numerous other chemokines are involved or differentially expressed. 829 For example the Fas and FasL systems are only weakly expressed in erythema multiforme compared with toxic epidermal necrolysis, 825 so that presumably this apoptotic pathway does not play a pivotal role in erythema multiforme. Likewise, there are fewer CD40L+ cells in erythema multiforme. 825 The finding of autoantibodies against desmoplakin I and II is probably an epiphenomenon.830.831. and 832.
The value of systemic corticosteroids in the treatment of erythema multiforme is hotly debated. Some relief of systemic symptoms is achieved but there is no evidence that their use improves the overall mortality or long-term morbidity. 833 Control with thalidomide has been achieved. The role of antivirals is controversial but prophylactic antiviral therapy with acyclovir (aciclovir) or valacyclovir (valaciclovir) inhibits HSV reactivation in recurrent cases and prevents HAEM development. 828 Recurrent erythema multiforme/Stevens–Johnson syndrome has responded to mycophenolate mofetil. 833 Dapsone, interferon-α, 834 and intravenous immunoglobulins combined with corticosteroids have all been successful in the treatment of some cases.
In established lesions, there is a lichenoid (interface) reaction pattern with a mild to moderate infiltrate of lymphocytes, some of which move into the basal layer, thereby obscuring the dermoepidermal interface (Fig. 3.19). Some of the intraepidermal lymphocytes are of the large granular subtype. 837 This is associated with prominent epidermal cell death, which is not confined to the basal layer. Apoptosis is the mechanism of cell death. 838 There is also basal vacuolar change and some epidermal spongiosis. 835 One study has found that an acrosyringeal concentration of apoptotic keratinocytes in erythema multiforme is a clue to a drug etiology. These changes are likely to be accompanied by an inflammatory infiltrate containing eosinophils. 839
Erythema multiforme. (A) An early case from the dorsum of the hand. (B) A more established case in which cell death involves keratinocytes within and above the basal layer. The infiltrate of lymphocytes tends to obscure the dermoepidermal interface. (H & E)
Vesicular lesions are characterized by clefting at the dermoepidermal junction and prominent epidermal cell death in the overlying roof (Fig. 3.20). This may involve single cells or groups of cells, or take the form of confluent necrosis.
Erythema multiforme with early subepidermal vesiculation. Preservation of the basket-weave pattern of the stratum corneum is a characteristic feature. (H & E)
The dermal infiltrate in erythema multiforme is composed of lymphocytes and a few macrophages involving the superficial and mid-dermal vessels and a more dispersed infiltrate along and within the basal layer. In severe cases of erythema multiforme showing overlap features with toxic epidermal necrolysis (see below), the infiltrate may be quite sparse with confluent necrosis of the detached overlying epidermis. Eosinophils are not usually prominent in erythema multiforme, although they have been specifically mentioned as an important feature in some reports.835.840.841. and 842. We have seen a case with numerous dermal eosinophils with eosinophilic microabscesses in the epidermis. 843 Likewise, a vasculitis has been noted by some, 844 but specifically excluded by most.835.836. and 845. Nuclear dusting, not related to blood vessels, is sometimes present. 846
Erythema multiforme has been divided in the past into epidermal, dermal, and mixed types based on the corresponding predominant histological features. 846 In the epidermal type there was prominent epidermal damage. In the dermal type there was pronounced dermal papillary edema leading to subepidermal vesiculation; some basal epidermal damage was to be seen in some areas of the biopsy. It seems likely that diseases other than erythema multiforme – severe urticarias and urticarial vasculitis – have been included in this category of dermal erythema multiforme. There is little merit in the continued separation of these three histological subtypes. 847 The rare cases with subcorneal pustules are probably not variants of erythema multiforme as reported. 848
Erythema multiforme-like changes can be seen in some biopsies of paraneoplastic pemphigus (see p. 151). Usually, foci of suprabasal acantholysis will be seen in some areas of the biopsy. Distinctive immunofluorescence changes are also present in paraneoplastic pemphigus.
Erythema multiforme-like changes can be seen in biopsies taken from the hypersensitivity reactions to phenytoin, carbamazepine and related drugs. Interestingly, the clinical picture does not resemble erythema multiforme.
Direct immunofluorescence shows intraepidermal cytoid bodies, representing degenerate keratinocytes, which stain in a homogeneous pattern usually with IgM and sometimes C3. 849 Frequently, there is granular staining for C3 along the dermoepidermal junction and, in early lesions, also in papillary dermal vessels.850. and 851. The presence of properdin suggests activation of the alternate complement pathway. 851 Matrix metalloproteinases 2, 9, and 11 are expressed in erythema multiforme. 852
Toxic epidermal necrolysis (TEN), regarded as the most severe form of an erythema multiforme spectrum, presents with generalized tender erythema which rapidly progresses into a blistering phase with extensive shedding of skin.851.853.854.855.856.857.858.859. and 860. Erosive mucosal lesions are usually present. Multiple intestinal ulcers have also been reported. 809 The mortality approaches 35%.860. and 861. The risk of death can be predicted from the quantitative ‘severity of illness’ score (SCORTEN). 862 However, respiratory involvement in TEN portends a poor prognosis that may not be reflected in SCORTEN. 863 The extent of the necrolysis (skin shedding) is one of the principal prognostic factors, and a classification system based on the extent of epidermal detachment has therefore been proposed:701. and 864.
1. Stevens–Johnson syndrome – mucosal erosions and epidermal detachment below 10% of total body area;
2. Stevens–Johnson syndrome/toxic epidermal necrolysis overlap (SJS/TEN) – epidermal detachment between 10 and 30%;
3. toxic epidermal necrolysis – epidermal detachment more than 30%.
Drugs are incriminated in the etiology in the majority of cases, 865 particularly sulfonamides, 866 anticonvulsants,867.868.869.870. and 871. selective serotonin reuptake inhibitors (SSRIs), 872 and non-steroidal anti-inflammatory drugs, such as phenylbutazone, isoxicam, and piroxicam.873. and 874. The incidence of TEN in adults secondary to trimethoprim-sulfamethoxazole has been calculated to be 2.6 per 100 000 exposures, while in patients who are HIV-positive, the rate is 8.4 cases per 100 000 exposures. 875 Specific drugs implicated include allopurinol,806.876.877. and 878. ranitidine,879. and 880. clindamycin, 881 telithromycin, 882 clarithromycin,883. and 884. moxifloxacin, 885 isoniazid, 871 thiacetazone, 871 chloroquine, 886 hydroxychloroquine,887. and 888. amphetamines (including ‘speed’),889. and 890. tetrazepam,891. and 892. famotidine, 893 indapamide, 894 clobazam, 895 oxaprozin, 896 pseudoephedrine, 897 diacerein, 898 methotrexate,899. and 900. leflunomide, 901 lansoprazole,902. and 903. cytosine arabinoside, 904 lamotrigine,905.906. and 907. voriconazole, 875 rituximab, 875 imatinib, 875 carbamazepine,907.908. and 909. latanoprost eye drops (with timolol and dorzolamide in one case), 910 trimethoprim-sulfamethoxasole,911. and 912. ceftazidime, 913 vancomycin,913. and 914. ciprofloxacin, 915 colchicine, 916 acetaminophen (paracetamol), 917 nevirapine,918. and 919. the SSRIs (fluvoxamine, paroxetine, fluoxetine, sertraline, citalopram, escitalopram), 872 mefloquine, 801 mifepristone/gemeprost, 920 alfuzosin, 921 oxazepam, 922 ritodrine/indomethacin/betamethasone therapy for preterm labor, 923 ampicillin/sulbactam, 924 phenytoin, 870 zonisamide associated with reactivation of HHV-6, 925 and ethambutol. 870
Another association of toxic epidermal necrolysis includes angioimmunoblastic T-cell lymphoma. 926
The pathogenesis is uncertain but it appears that most patients with toxic epidermal necrolysis have an abnormal metabolism of the offending drug, which leads to an increased production of reactive metabolites.701. and 868. Some genetic susceptibility is suggested by the increased incidence of HLA-B12 in affected individuals.927. and 928. It has developed in a mother and her 22-week-old fetus, 929 and in a premature infant. 930
Epidermal necrosis is probably mediated by cytokines from drug-specific cytotoxic T lymphocytes, 931 such as tumor necrosis factor-α,757.826.932. and 933. but it is unlikely this is the only mediator of epidermal damage (see below). 934 Apoptosis of keratinocytes also occurs. 935 It is thought that this may result from the interaction between the death receptor Fas (CD95) and its ligand present on epidermal cells. 936 The action of cytokines would explain the apparent discrepancy between the extent of the damage and the paucity of the dermal infiltrate.
The majority of non-keratinocytic cells in the epidermis are CD8+ lymphocytes and macrophages, while the lymphocytes in the papillary dermis are CD4+.937.938. and 939. Even though the cell-mediated immune response is characterized, in part, by a Th1 profile, there is increasing evidence in favor of the development of a Th2-mediated response in SJS/TEN, whereas erythema multiforme is characterized by a dominant Th1 profile.940. and 941. Drug-specific CD8+ T lymphocytes appear to be the main triggering agents of the massive epidermal damage in SJS/TEN by secretion of perforin, granzyme B, and cytokines such as TNF-α.825.928. and 942. Furthermore, the interaction between Fas and Fas ligand triggering the caspase cascade is another mechanism of apoptosis, but it may not be the critical mediator of cell death as once thought. 934 The CD40/CD40L pathway is yet another mechanism of cell damage. 943 Serum interleukin-13 (IL-13) levels are also increased in SJS/TEN but not in erythema multiforme. 940 There are elevated levels of IL-10, IL-6, IL-8, IL-2 receptor, and TNF-α in blister fluid.944.945. and 946. The sera from patients with TEN contain autoantibodies to periplakin which may play a role in the pathogenesis as a humoral autoimmune mechanism. 947 Antidesmoplakin autoantibodies also circulate. 948
Patients on corticosteroid therapy may still develop toxic epidermal necrolysis; corticosteroids may delay the onset of the disease, but not halt its progression.949. and 950. Patients infected with the human immunodeficiency virus may develop toxic epidermal necrolysis similar to immunocompetent patients.951.952. and 953.
As severe Stevens–Johnson syndrome (SJS) forms part of a spectrum with toxic epidermal necrolysis, the following comments, in most cases, apply to both diseases. Prompt withdrawal of the offending drug should be a priority. Doing so reduces the risk of death by about 30% per day. 954 Referral to a burns unit or specialized dermatology unit are generally agreed upon steps. Evidence for and against the use of intravenous immunoglobulins and systemic corticosteroids exists,875.922.924. and 955. but combination therapies may be of value.928. and 942. A recent observational study from France and Germany found no evidence of a benefit for any specific treatment compared with supportive care. 936 However, the report concluded that ‘the trend for a beneficial effect of corticosteroids deserves further exploration’. 936 Intravenous immunoglobulins were originally used because they block Fas ligand binding to the Fas receptor, which at one time was thought to be the most important cause of apoptosis in this disease.956.957.958.959. and 960. On a similar basis, anti-TNF-α therapy with infliximab, 961 and the use of N-acetylcysteine, and pentoxifylline have also been tried.962. and 963. Intravenous cyclosporine has also been used. 890 Unfortunately placebo-controlled trials are logistically difficult to organize. One such trial in the past using thalidomide had to be halted when significant mortality occurred. 963
Nursing care is important in the management of these patients. Secondary infections, some fatal, occur. 964 Wound debridement is important. 960 The use of skin substitutes is often advocated. 965 Re-epithelialization may be delayed in patients with hyperbilirubinemia. 966 A treatment protocol has been developed by the University of Florida. 960
There is a subepidermal bulla with overlying confluent necrosis of the epidermis and a sparse perivascular infiltrate of lymphocytes (Fig. 3.21). In early lesions there is some individual cell necrosis which may take the form of lymphocyte-associated apoptosis (satellite cell necrosis) with an adjacent lymphocyte or macrophage. This has been likened to the changes of graft-versus-host disease (GVHD; see below).854. and 968. In established lesions of toxic epidermal necrolysis there is full thickness epidermal necrosis which is not seen in GVHD. 968 A more prominent dermal infiltrate is seen in those cases which overlap with erythema multiforme. If the degree of dermal mononuclear inflammation is quantified, some prognostic information, equivalent to that provided by SCORTEN, can be obtained. 969 In one study of 37 patients, 73% of patients (n = 11) with sparse inflammation survived, but only 47% (n = 7) with moderate and 29% (n = 2) with extensive inflammation survived. 969 Sweat ducts show a variety of changes ranging from basal cell apoptosis to necrosis of the duct. 970 In the healing phase, milia and disturbances of pigmentation are common. Scarring and keloids may also develop. Verrucous hyperplasia of the epidermis is a rare response. 971 Vulval and vaginal adenosis are other late responses.972. and 973.
Toxic epidermal necrolysis. There is a subepidermal cell-poor blister with epidermal necrosis in the roof. (H & E)
A diffuse deposition of immunoreactants has been found in the mid-epidermis on immunofluorescence. 974
Graft-versus-host disease (GVHD) is a systemic syndrome with important cutaneous manifestations. It is usually seen in patients receiving allogeneic immunocompetent lymphocytes in the course of bone marrow transplants used in the treatment of aplastic anemia, 975 leukemia, or in immunodeficiency states.739.976.977.978.979. and 980. Acute GVHD develops in approximately one-third of HLA-matched recipients of allogeneic bone marrow.981. and 982. It also follows stem cell transplantation. 983 Risk factors for the development of acute cutaneous GVHD after allogeneic stem cell transplantation include a diagnosis of chronic myeloid leukemia, HLA disparity, and conditioning regimens such as total body irradiation. 983 It may also occur following maternofetal blood transfusions in utero, 984 intrauterine exchange transfusions and the administration of non-irradiated blood products to patients with disseminated malignancy and a depressed immune system.985.986.987.988.989.990. and 991. It is seen only rarely after solid organ transplantation.992. and 993. It is also less common with cord-blood than with bone marrow transplants in children receiving either product from HLA-identical siblings. 994 Rarely, immunocompetent patients are at risk, particularly those subject to cardiac surgery.995.996. and 997. The acute stage can be precipitated in some individuals by autologous and syngeneic bone marrow transplantation;998.999. and 1000. chronic lesions (see below), particularly lichenoid ones, are rare. 1001 Acute GVHD superimposed on pre-existing lichenoid chronic GVHD has been reported; it followed reinduction chemotherapy. 1002 GVHD has been reported in patients with a thymoma or lymphoma.1003.1004.1005. and 1006. Sclerodermoid GVHD-like lesions have also developed long after the clinical resolution of drug-induced hypersensitivity syndrome. 1007
There is an early acute phase with vomiting, diarrhea, hepatic manifestations, and an erythematous macular rash.978.1008. and 1009. Rarely, it is confined to the flexures. 1010 Uncommonly, there are follicular papules1011 or blisters; 1012 rarely, toxic epidermal necrolysis ensues.978. and 1013. Just two erythematous nodules were the presenting features in one case. 1014 A pustular acral erythema with associated eccrine squamous syringometaplasia has also been reported. 1015 Ichthyosiform features may occur in both the acute and chronic forms.1016. and 1017. Localization to an area of skin affected by piebaldism, and in another case herpes zoster, has been reported.1018. and 1019. Lichenoid nail changes also occur. 1020 They usually persist in chronic disease. 1021 The chronic stage develops some months or more after the transplant. A preceding acute stage is present in 80% of these patients.978. and 1022. Chronic GVHD has an early lichenoid phase which resembles lichen planus and includes oral lesions.1023. and 1024. Linear lichenoid lesions have been reported both in a dermatomal distribution and following Blaschko’s lines.1025.1026.1027.1028. and 1029. Rarely, dermatomal lesions occur at sites of varicella-zoster infection.1029. and 1030. A poikilodermatous phase may precede the eventual sclerodermoid phase. The lesions of the latter may be localized1031 or generalized.978. and 1032. Sclerodermatous GVHD (see p. 312) has a prevalence of approximately 3% in patients receiving allogeneic bone marrow transplants. 1033 Other late manifestations include alopecia, a lupus erythematosus-like eruption, cicatrizing conjunctivitis, 1034 pyogenic granuloma and angiomatous lesions, 1035 wasting, diffuse melanoderma, 1036 leukoderma and leukotrichia, 1037 esophagitis, liver disease, and the sicca syndrome. Acute GVHD may be a late manifestation (>100 days after transplantation), following the suspension or tapering of immunosuppressive drugs. It can be seen after traditional transplants and the newer non-myeloablative technique. 1038 Late-onset acute GVHD is a predictor of chronic GVHD. 1038
The pathogenesis appears to be complex, 1039 but the essential factor is the interaction of donor cytotoxic T lymphocytes with recipient minor histocompatibility antigens.1040.1041. and 1042. Several effector cell populations appear to be involved.1043. and 1044. Recent work suggests that this is an over-simplistic explanation of the pathogenesis. 1045 Not only is direct cellular cytotoxicity involved, but also soluble mediators such as IFN-γ, CXCR3, TNF-α, FasL, and TNF-related apoptosis-inducing ligand (TRAIL) play a significant role in the pathogenesis of acute GVHD.1045. and 1046. Young rete ridge keratinocytes1047 and Langerhans cells1042. and 1048. are preferred targets. The acute stage is associated with HLA-DR expression of keratinocytes.1049.1050. and 1051. Factor XIIIa-positive dermal dendrocytes appear to play some role,1052. and 1053. possibly in the regulation of the connective tissue remodeling that follows epidermal destruction. Recent work suggests that HHV-6 reactivation may play a role in the pathogenesis of rash/GVHD after allogeneic stem cell transplantation. 1054
The treatment of acute GVHD is beyond the scope of this book, involving as it does a disease in which the cutaneous manifestations may be a small component of a systemic disease. Chronic cutaneous GVHD has been treated with corticosteroids and immunosuppressants such as cyclosporine. They have had a limited effect on the disease, not to mention the long-term effects of corticosteroids. Chronic disease may also be treated by extracorporeal photopheresis (ECP). 1055 It also induces, as a side effect, immediate and progressive apoptosis. 1056 A consensus statement on its use was published in 2008. 1055 UVA1 phototherapy, 1057 tacrolimus ointment, 1058 and pimecrolimus1059 have also been used in chronic cutaneous GVHD.
In early acute lesions there is a sparse superficial perivascular lymphocytic infiltrate with exocytosis of some inflammatory cells into the epidermis. The number of these cells correlates positively with the probability of developing more severe, acute GVHD.1061. and 1062. The infiltrate in GVHD developing after solid organ transplantation is usually brisk in comparison to the more sparse inflammation following bone marrow transplantation. 1063 This infiltrate is accompanied by basal vacuolation. Established lesions are characterized by more extensive vacuolation and lymphocytic infiltration of the dermis and scattered, shrunken, eosinophilic keratinocytes with pyknotic nuclei, at all levels of the epidermis. 978 These damaged cells are often accompanied by two or more lymphocytes, producing the picture known as ‘satellite cell necrosis’ (lymphocyte-associated apoptosis) (Fig. 3.22). 1064 A similar picture is sometimes seen in subacute radiation dermatitis (see p. 528), 1065 and in the cutaneous eruption of lymphocyte recovery (see below). There is a lack of specificity in skin biopsy specimens taken in the initial 3 weeks after bone marrow transplantation. 1066 Distinction from a drug eruption is also difficult.1067.1068. and 1069. The presence of eosinophils is generally taken to favor a drug reaction, but this is not a correct assumption as eosinophils are occasionally seen in GVHD. 1070 Marra et al have highlighted the perils of using skin biopsy specimens to distinguish between drug reactions and cutaneous GVHD. 1071 They reported three cases in whom the presence of eosinophils on skin biopsy led to the mistaken diagnosis of a drug reaction, leading to a delay in treatment for their GVHD. 1071 Others have questioned the value of skin biopsy in this condition. 1072 The presence of more than five apoptotic keratinocytes, predominantly involving adnexal keratinocytes, is said to favor GVHD. Fluorescent in-situ hybridization analysis for donor lymphocytes in a skin biopsy specimen can serve as an early diagnostic tool for GVHD. 1073 Fulminant lesions in the acute stage resemble those seen in toxic epidermal necrolysis with subepidermal clefting and full thickness epidermal necrosis. Follicular wall necrosis was reported in one case. 1014 These various changes are often graded as to severity. 1074 However, the biopsy findings after bone marrow transplantation correlate poorly with the clinical severity of the skin rash and in predicting progression of the disease to a more severe clinical state.1075. and 1076. Even normal-appearing skin is not necessarily normal on histological examination. 1077 The following scheme has been proposed by Horn: 1078
(A) Graft-versus-host disease. (B) Lymphocytes are in close apposition to apoptotic keratinocytes. (H & E)
Omenn syndrome (OMIM 603554), an autosomal recessive form of severe combined immunodeficiency due to mutations in the RAG1, RAG2, or Artemis genes, presents soon after birth with erythroderma, desquamation, recurrent infections, hepatosplenomegaly, and failure to thrive. B lymphocytes are usually absent, but the T lymphocytes in the peripheral blood that are activated and oligoclonal can cause a GVHD, the reason for its mention here. Skin biopsies in Omenn syndrome look similar to those in GVHD. However, in Omenn syndrome there is always acanthosis and parakeratosis, whereas in GVHD the epidermis is generally flat, rarely with parakeratosis. 1079 Inflammation is more marked in Omenn syndrome. 1079
In the early chronic phase of GVHD, the lichenoid lesions closely resemble those of lichen planus, although the infiltrate is not usually as dense. 1080 Sometimes the pattern may even resemble that seen in acute GVHD. 1081 Pigment incontinence may be prominent. Biopsies taken from follicular papules resemble lichen planopilaris.1011. and 1023. A rare manifestation is so-called ‘columnar epidermal necrosis’ characterized by small foci of total epidermal necrosis accompanied by a lichenoid tissue reaction. 1082 Immunofluorescence shows a small amount of IgM and C3 in colloid bodies in the papillary dermis and some immunoglobulins on necrotic keratinocytes.1023. and 1024.
In the late sclerodermoid phase, there are mild epidermal changes such as atrophy and basal vacuolation. There is thickening of dermal collagen bundles which assume a parallel arrangement. The dermal fibrosis, which may result in atrophy of skin appendages, usually extends into the subcutis, resulting in septal hyalinization. 1060 Subepidermal bullae were present in one reported case. 1083
The recently introduced immunomodulatory agent roquinimex has produced eccrine sweat gland necrosis in a number of instances. 1084
Although in its infancy, composite tissue allografts are being performed with increasing frequency. One such example is the hand allograft. Rejection of allografted skin manifests with changes that are characteristic but not very specific. 1085 An excellent review of this topic was published in 2008 by Kanitakis. 1085 The changes range from mild morbilliform reactions, to spongiotic vesiculation, to a pseudolymphomatous pattern ± lichenoid changes, to a severe necrotizing (grade IV) pattern. 1085
Ultrastructural examination shows ‘satellite cell necrosis’ in both stages with lymphocytes in close contact with occasional keratinocytes, 1086 some of which show the changes of apoptosis. The term ‘lymphocyte-associated apoptosis’ is therefore more appropriate than ‘satellite cell necrosis’. 1087 Lymphocytes are also in contact with melanocytes1088 and Langerhans cells, the latter being reduced in number. Melanosomes may be increased in the melanocytes. The late sclerotic phase is distinct from scleroderma, with some apoptotic cells in the epidermis and numerous active fibroblasts in the upper dermis. 1089
The original description of this ‘entity’ involved patients who developed a maculopapular eruption after receiving cytoreductive therapy (without bone marrow transplant) for acute myelogenous leukemia. 1090 The lesions usually developed 14–21 days later, coincident with the return of lymphocytes to the circulation. 1078 The histological similarities between this eruption and those seen with mild GVHD and with the administration of cyclosporin A (cyclosporine) led to the suggestion that all three conditions represent variations on the theme of lymphocyte recovery. 1078
The eruption is characterized by an upper dermal perivascular infiltrate of small T lymphocytes with accompanying vascular dilatation. There is mild exocytosis of lymphocytes with occasional apoptotic keratinocytes. Lymphocytes are sometimes seen in apposition with these degenerate cells (‘satellite cell necrosis’). The appearances resemble mild GVHD. 1091 In one report, Pautrier-like microabscesses containing CD4+ cells were present in the epidermis, mimicking mycosis fungoides. 1092
The systemic administration of recombinant cytokines prior to marrow recovery leads to a relatively heavy lymphocytic infiltrate with nuclear pleomorphism and hyperchromasia. 1093
A lichenoid reaction pattern with interface changes resembling those seen in erythema multiforme or fixed drug eruptions has been reported in some patients with the acquired immunodeficiency syndrome and in whom the clinical presentation suggested a drug reaction. 1094 The patients had numerous opportunistic infections and all had received at least one medication prior to the onset of the rash. It has been suggested that systemic and cutaneous immune abnormalities may be relevant in the pathogenesis. 1094
There is little justification for the continuation of this diagnosis as a discrete entity.
Lupus erythematosus is a chronic inflammatory disease of unknown etiology which principally affects middle-aged women. It has traditionally been regarded as an immune disorder of connective tissue, along with scleroderma and dermatomyositis. However, a striking feature of cutaneous biopsies is the presence in most cases of the lichenoid reaction pattern (interface dermatitis). It may not be present in tumid forms, in lupus profundus (panniculitis) and in lymphocytic infiltration of the skin, if indeed this is truly a variant of lupus erythematosus.
Three major clinical variants are recognized – chronic discoid lupus erythematosus which involves only the skin, systemic lupus erythematosus which is a multisystem disease, and subacute lupus erythematosus in which distinct cutaneous lesions are sometimes associated with mild systemic illness.1095. and 1096. Some overlap exists between the histological changes seen in these various clinical subsets. 1097 There are several less common clinical variants which will be considered after a discussion of the major types.
A recent addition to these subsets is undifferentiated connective tissue disease, also called latent or incomplete lupus, in which signs and symptoms do not fulfill any of the accepted classification criteria for the various named connective tissue diseases.1098. and 1099.
The typical lesions of discoid lupus erythematosus (DLE) are sharply demarcated, erythematous, scaly patches with follicular plugging. They usually involve the skin of the face, often in a butterfly distribution on the cheeks and bridge of the nose. The neck, scalp, eyelids,1100.1101.1102. and 1103. lips, 1104 oral mucosa,1105. and 1106. and hands, including the nails, 1107 are sometimes involved. Periorbital localization has been reported. 1108 There is a female preponderance. 1109 The lesions may undergo atrophy and scarring. Acneiform pitting scars are a rare presentation of DLE. 1110 Cicatricial alopecia may result from scalp involvement.1111. and 1112. DLE is rare in children.1113.1114.1115.1116.1117.1118. and 1119. Less than 2% of patients with DLE have an onset before 10 years of age. 1120 Children have a particularly high level of transition to systemic disease. 1121 Squamous cell carcinoma is a rare complication in any site.1122.1123. and 1124. Reflectance confocal microscopy awaits further evaluation as a diagnostic tool for DLE, although it appears to be promising for biopsy site selection. 1125
A hypertrophic variant of DLE in which verrucous lesions develop, usually on the arms, has been reported.1126.1127. and 1128. Lupus erythematosus hypertrophicus et profundus is a very rare destructive variant of hypertrophic DLE with a verrucous surface and eventual subcutaneous necrosis. 1129 The face and arms are the most common sites of hypertrophic DLE. Lesions resembling keratoacanthomas may develop. 1130 Verrucous lesions were present in one patient with lupus erythematosus associated with porphyria. 1131 This variant of lupus erythematosus may be misdiagnosed as squamous cell carcinoma on superficial shave biopsy. 1132 Squamous cell carcinoma is an uncommon late complication. 1128
Annular lesions, resembling erythema multiforme, may rarely develop acutely in patients with all forms of lupus erythematosus.1133.1134. and 1135. This syndrome, known as Rowell’s syndrome, is also characterized by a positive test for rheumatoid factor and speckled antinuclear antibodies.1133.1136.1137.1138.1139.1140. and 1141. The antiphospholipid syndrome may also be present.1142. and 1143. Lupus-associated toxic epidermal necrolysis may represent a more severe variant of Rowell’s syndrome. 1144
Papulonodular lesions associated with diffuse dermal mucin are uncommon manifestations of chronic cutaneous lupus erythematosus.1145. and 1146. This mucinosis has presented as periorbital edema. 1147 This variant is part of the spectrum of tumid lupus erythematosus (see below).
Tumid lupus erythematosus (lupus erythematosus tumidus) 1148 consists of erythematous, urticaria-like, non-scarring plaques and sometimes papules on the face, neck, and upper trunk. They are usually in sun-exposed areas.1149. and 1150. Monolateral severe eyelid erythema and edema are unique manifestations of this variant. 1151 Lesions may have a fine scale and be pruritic. 1152 It usually occurs in a setting of DLE but sometimes systemic lupus erythematosus has been present or rarely develops subsequently.1153. and 1154. Tumid lupus has also developed following the use of highly active antiretroviral therapy (HAART) for HIV infection. 1155 Tumid lupus is part of a spectrum that includes the papulonodular type (see above). A study of 80 patients with this disease concluded that on the basis of specific histopathological features, this condition should be considered a separate entity of cutaneous lupus erythematosus. 1156 There is usually a good response to antimalarials with this form of the disease. 1149
Lymphocytic infiltration of the skin (of Jessner and Kanof) is now regarded as a variant of DLE,1157. and 1158. although there is still speculation that some cases may represent borreliosis. 1159 Although various studies in the 1980s concluded that they were separate entities on the basis of the direct immunofluorescence, the usual absence of epidermal damage and the phenotype of the infiltrating lymphocytes, recent studies have suggested that it may be part of the tumid spectrum of DLE. Provocative phototesting gives similar results. 1158 A recent comparison of the histopathological and clinical features of this condition and tumid lupus erythematosus showed more similarities than differences, supporting a continuous spectrum of these two conditions. 1160
Linear lesions, often following the lines of Blaschko, have been reported.1161.1162.1163.1164. and 1165. Many of them have been on the face,1166.1167. and 1168. although the trunk has also been involved. 1169 Several cases of a sclerodermiform linear lupus erythematosus have been reported. 1170 It may represent an unusual mosaicism along Blaschko’s lines or the transfer of microchimerisms that mount a chronic graft-versus-host-like reaction. 1170
Discoid lesions may be seen in up to 20% of individuals with systemic lupus erythematosus, 1171 often as a presenting manifestation. 1117 It is therefore difficult to estimate accurately the incidence of the progression of the discoid to the systemic form. This is in the order of 5–10%1172.1173. and 1174. and is most likely in those who present abnormal laboratory findings, such as a high titer of antinuclear antibody (ANA) and antibodies to DNA, from the beginning of their illness. 1175 Approximately 70% of patients possess low titers of anti-Ro/SSA antibodies and it remains to be seen whether such patients are at greater risk of progression to the systemic form.1174.1176. and 1177. Some patients with localized lesions may progress to more widespread disease. 1178 Visceral manifestations are absent in uncomplicated DLE. An increased incidence of various haplotypes has been found;1179. and 1180. HLA-DRB1 alleles are involved in the genetic susceptibility of a Mexican population. 1181 It has been suggested that genes encoding immunoregulatory molecules may determine individual susceptibility to lupus erythematosus. 1182 Lesions resembling discoid or subacute lupus erythematosus can be found in the female carriers of X-linked chronic granulomatous disease,1183.1184.1185.1186.1187. and 1188. and rarely in an autosomal form of that disease.1189. and 1190. DLE has also been reported in association with Cockayne’s syndrome, 1191 with a deficiency of C5, 1192 C2, 1193 and other immunodeficiency syndromes. 1194 A combination of complement deficiency and smoking may be a risk factor for cutaneous lupus erythematosus in men. 1195 It has also been induced by adalumimab, 1196 infliximab, 1197 pantoprazole, 1198 and by cyclosporine. 148 Phototesting with UVA or combined UVA and UVB irradiation will produce positive reactions in about half of all patients tested. 1199
The inflammatory infiltrate in cutaneous lupus erythematosus is composed of T lymphocytes, with a slight predominance of CD4+ over CD8+ cells. In DLE, type I IFNs and potentially autoreactive cytotoxic lymphocytes targeting adnexal structures are highly associated with scarring lesions. 1200 There is a strong expression of granzyme B and the type I interferon-induced protein MxA. 1200
The treatment of cutaneous lupus erythematosus varies with the severity of the disease, the number of lesions, their localization, the subtype of the disease, and the presence of photosensitivity. Approximately half of all patients with chronic cutaneous lupus erythematosus have lesions limited to the head, the neck or both. These patients differ from those with widespread DLE by less often having a positive ANA titer, and having much less likelihood of progression to systemic disease. 1201 They can be treated with sunscreens and topical corticosteroids of low to medium potency. Oral antimalarials or retinoids can be used when topical agents are not effective.1112. and 1120. Systemic corticosteroids are rarely effective for DLE.1201. and 1202. Patients with the hypertrophic form usually require intralesional injections of triamcinolone with or without oral retinoids. Patients with tumid lupus are responsive to antimalarials. 1201 They also require sun protection. 1201 Topical or intralesional corticosteroids have been used as adjunctive therapies. 1202 Topical calcineurin inhibitors such as tacrolimus ointment 0.1% and pimecrolimus cream have been used in refractory cases. 1203 They are of no use in hypertrophic lesions. 1204 Efalizumab (directed against CD11a) has been used in DLE. 1205 Only small numbers have been treated with these newer immunomodulators and further trials are needed. Finally, UVB hardening has been used in patients with photosensitive disease of all types. It may lead to improved tolerance for environmental ultraviolet radiation. 1206
Discoid lupus erythematosus is characterized by a lichenoid reaction pattern and a superficial and deep dermal infiltrate of inflammatory cells which have a tendency to accumulate around the pilosebaceous follicles (Fig. 3.23). In scalp lesions with scarring alopecia, there is considerable reduction in the size of sebaceous glands and the lymphocytic infiltrate is maximal around the mid-follicle at the level of the sebaceous gland. 1111 The lichenoid reaction (interface dermatitis) takes the form of vacuolar change (‘liquefaction degeneration’), although there are always scattered Civatte bodies (apoptotic keratinocytes). In lesions away from the face the number of Civatte bodies is always much greater and a few colloid bodies may be found in the papillary dermis (Fig. 3.24). In older lesions, there is progressive thickening of the basement membrane, which is best seen with a PAS stain. Other epidermal changes include hyperkeratosis, keratotic follicular plugging and some atrophy of the malpighian layer. 1208
Discoid lupus erythematosus. The dermal infiltrate is both superficial and deep with perifollicular accentuation. (H & E)
Discoid lupus erythematosus. There is more cell death and less vacuolar change in the basal layer than usual. Distinction from the subacute form is difficult in these cases. (H & E)
The dermal infiltrate is composed predominantly of lymphocytes with a few macrophages. Atypical lymphocytes, mimicking mycosis fungoides, have been reported in one case. 1209 Occasionally there are a few plasma cells and rarely there are neutrophils and nuclear dust in the superficial dermis in active lesions. Plasma cells are prominent in oral lesions. 1105 Fibrin extravasation and superficial edema are also seen in the papillary dermis in some early lesions. Mucin is sometimes increased, but only rarely are there massive amounts. 1210 Amyloid of keratinocyte origin1211 and calcification have been reported on a few occasions.1212. and 1213.
In tumid lesions there is increased dermal mucin in all cases, often accompanied by subepidermal edema (Fig. 3.25). Some cases have only a sparse inflammatory cell infiltrate, while others have a heavy infiltrate of lymphocytes and less mucin. 1214 A few scattered neutrophils may be present. Epidermal involvement is uncommon;1152. and 1215. in one study of 80 cases, epidermal atrophy and alterations at the dermoepidermal junction were absent in all cases. 1156 In a blinded comparison of tumid lupus and Jessner’s lymphocytic infiltrate there were only slight differences between the two. 1160 Slight epidermal atrophy and focal thickening of the dermoepidermal junction were more common in tumid lupus and the lymphocytic infiltrate was less dense in tumid lupus than in Jessner’s lymphocytic infiltrate, supporting a continuous spectrum for these two disorders. 1160 A pattern resembling that seen in tumid lupus erythematosus has been reported at the injection site of interferon. 1216 There was abundant dermal mucin in addition to the heavy lymphocytic infiltrate along hair follicles with vacuolar change of the basal layer of the follicles. 1216 A similar, but less severe reaction has since been reported. 1217
Discoid lupus of tumid type. (A) There is a superficial and deep dermal infiltrate (H & E). (B) Dermal mucin is greatly increased (Alcian blue). (C) Another case which might have been called Jessner’s lymphocytic infiltrate in the past. (H & E)
In hypertrophic lesions there is prominent hyperkeratosis and epidermal hyperplasia. There may be a vague resemblance to a superficial squamous cell carcinoma, particularly on shave biopsy.1218. and 1219. Elastic fibers are often present between epidermal cells at the tips of the epidermal downgrowths. Transepidermal elimination of these fibers also occurs. 1128
Direct immunofluorescence of involved skin in discoid lupus will show the deposition of immunoglobulins, particularly IgG and IgM, along the basement membrane zone in 50–90% of cases (Fig. 3.26).1220.1221.1222.1223.1224.1225. and 1226. The incidence is less than 50% in the author’s experience; perhaps this is a reflection of subtropical cases in Caucasians or the prompt biopsy of younger lesions (see p. 63). Complement components are present less frequently. This so-called ‘lupus band test’ is positive much less often in lesions from the trunk. 1227 A positive lupus band test should always be interpreted in conjunction with the clinical and histological findings, 1228 as it may be obtained in chronic light-exposed skin1229 and some other conditions. 1230 Cytoid bodies, sometimes perifollicular in location, are present in more than half the cases. 1231
Discoid lupus erythematosus. A broad band of C3 is present along the basement membrane zone. (Direct immunofluorescence)
Subacute lupus erythematosus is characterized by recurring, photosensitive, non-scarring lesions which may be annular or papulosquamous in type.1233.1234. and 1235. They are widely distributed on the face, neck, upper trunk, and extensor surfaces of the arms. 1236 A case with only acral lesions has been reported. 1237 In another case arcuate plantar plaques were followed by a facial lesion. 1238 The patients frequently have a mild systemic illness with musculoskeletal complaints and serological abnormalities, but no central nervous system disease.1239. and 1240. Renal disease occurred in 16% of patients in one series. 1241 Severe visceral involvement is most uncommon. 1242 Cases with overlap features between the systemic and subacute forms have been reported.1243.1244. and 1245. Rare clinical presentations have included erythroderma,1246. and 1247. erythroderma and bullae, 1248 bullae alone, 1249 a poikilodermatous pattern, 1250 pityriasiform lesions, 1251 onset in childhood,1252. and 1253. the simultaneous occurrence of Sweet’s syndrome, 1254 and an association with the ingestion of thiazide, 1255 terbinafine,1256.1257.1258.1259. and 1260. etanercept, 1261 infliximab, 1262 leflunomide,1263. and 1264. bupropion, 1265 phenytoin, 1266 adalimumab, 1267 simvastatin, 1268 efalizumab, 1269 capecitabine, ticlopidine, 1270 anastrozole, 1271 antihistamines, 1272 calcium channel blockers, 1273 and griseofulvin. 1274 It has also been induced by radiation therapy1275 and contact with fertilizer- and pesticide-containing hay. 1276 It has followed the inhalation of tiotropium bromide, a bronchodilator. 1277 It has been reported in patients with hepatocellular carcinoma, 1278 prostate carcinoma, 1279 epidermoid carcinoma of the lung, 1280 and adenocarcinoma in situ of the esophagus. 1281 An interesting finding is that smokers with cutaneous lupus erythematosus are less responsive to antimalarial treatment than non-smokers. 1282 Drugs causing subacute lupus erythematosus are listed in Table 3.5.
The test for ANA is often negative if mouse liver is used as the test substrate, but positive if human Hep-2 cells are used. 1283 There is a high incidence of the anticytoplasmic antibody Ro/SSA;1284.1285.1286.1287.1288. and 1289. it is found in a higher incidence in those with annular rather than papulosquamous lesions. 1290 This antibody is also found in systemic lupus erythematosus, neonatal lupus erythematosus, in the lupus-like syndrome that may accompany homozygous C2 deficiency1291.1292.1293. and 1294. and in Sjögren’s syndrome.1295. and 1296. The Ro/SSA antigen is now known to be localized in the epidermis and it is thought that antibodies to this antigen are important in the initiation of tissue damage. 1297 Antigen expression is higher in photosensitive forms of lupus erythematosus. 1298 However, the antibody titer does not correlate with the activity of the skin disease. 1299
A drug etiology should first be excluded and any potential drugs should be ceased. Smokers seem to have more severe disease than non-smokers and this aspect requires attention. 1201 Depigmentation and other cosmetic concerns of the patient should also be addressed. Topical corticosteroids are usually used, the potency required depending on the area of the body affected.1201. and 1202. Patients apparently prefer creams over ointments, although ointments may be more potent. 1201 When lesions are not controlled with topical agents or intralesional corticosteroids, antimalarials such as hydroxychloroquine sulfate may prove effective. 1202 Thalidomide has also been used in these circumstances, but the number of patients treated is relatively small. 1300 Systemic corticosteroids are only ever partially effective for subacute lupus erythematosus. 1201 They should be restricted to patients with acute photosensitivity, and with vasculitis. Dapsone may also be of benefit in vasculitis. Topical immunomodulators such as tacrolimus and pimecrolimus can be used. 1203 Small numbers of cases have been treated with leflunomide, 1301 mycophenolate mofetil, 1302 and salbutamol cream. 1303 One case that was refractory to all treatments mentioned above, and also azathioprine and methotrexate, responded to efalizumab (the monoclonal antibody to CD11a). 1304
The histopathological features differ only in degree from those seen in discoid lupus.1239. and 1306. Usually there is more basal vacuolar change, epidermal atrophy and dermal edema and superficial mucin than in discoid lupus, but less hyperkeratosis, pilosebaceous atrophy, follicular plugging, basement membrane thickening and cellular infiltrate (Fig. 3.27).1305.1306.1307. and 1308. The pattern can be characterized as a pauci-inflammatory, vacuolar, lymphocytic interface dermatitis. 1214 Apoptotic keratinocytes (Civatte bodies) are sometimes quite prominent in subacute lupus erythematosus; they may be found at various levels within the epidermis, resembling erythema multiforme (Fig. 3.28).1309. and 1310. Furthermore, the infiltrate is usually confined more to the upper dermis than in discoid lupus.1305. and 1311. The above features relate to the more common annular form which may resemble erythema multiforme to varying degrees. 1141 The papulosquamous form of subacute lupus erythematosus has no distinguishing features to permit differentiation from the discoid form. 1307
(A) Subacute lupus erythematosus. (B) There is patchy basal vacuolar change and occasional Civatte bodies. (H & E)
Subacute lupus erythematosus. The infiltrate extends into the lower epidermis and cell death occurs at a slightly higher level than usual. This variant can be mistaken for erythema multiforme. (H & E)
The lupus band test (see above) shows immunoglobulins at the dermoepidermal junction in approximately 60% of cases. The band is usually not as thick or as intensely staining as in discoid lupus erythematosus. Very fine, dust-like particles of IgG (a speckled pattern) have been described predominantly, but not exclusively, in the cytoplasm of basal cells1311.1312.1313. and 1314. and also in the cellular infiltrate in the dermis. 1315 This pattern is not specific for subacute cutaneous lupus erythematosus or the presence of Ro/SSA antibodies. Dust-like particles were found in only 3% of cases in one recent study. 1316
In systemic lupus erythematosus (SLE) the changes in the skin are part of a much more widespread disorder. Four clinical manifestations are particularly important as criteria for the diagnosis of SLE: skin lesions, renal involvement, joint involvement, and serositis. 1095 The coexistence of the first two of these manifestations is sufficient to justify a strong presumption of the diagnosis.
Cutaneous lesions take the form of erythematous, slightly indurated patches with only a little scale. They are most common on the face, particularly the malar areas. The lesions are usually more extensive and less well defined than those of discoid lupus erythematosus and devoid of atrophy. Scarring is an important complication of all forms of lupus erythematosus. 1317 The lesions may spread to the chest and other parts of the body. In some instances, they may be urticarial, 1318 bullous,1319.1320. and 1321. follicular, 1322 mucinous, 1323 purpuric or, rarely, ulcerated. Facial edema is another presentation. 1324 It is important to remember that skin lesions do not develop at all in about 20% of patients with SLE; approximately the same proportion have discoid lesions of the type seen in chronic discoid lupus erythematosus, but usually without scarring. 1325 This latter group often has less severe disease. 1171 Subclinical inflammatory alopecia has also been reported. 1326 A spectrum of elastic tissue changes can occur in patients with lupus erythematosus. They range from mid-dermal elastolysis to anetoderma (see p. 345).
The digits, calves and heels are involved in the rare chilblain (perniotic) lupus which results from microvascular injury in the course of SLE.1327.1328. and 1329. A verrucous form of chilblain lupus has been reported in an adult. 1330 Red lunulae have been reported in association with chilblain lupus and also as an isolated phenomenon in SLE.1331.1332. and 1333. Familial chilblain lupus (OMIM 610448) is due to a mutation in the TREX1 gene on chromosome 3p21. It is allelic to Aicardi–Goutières syndrome (OMIM 225750), a rare genetic leukoencephalopathy. 1334
SLE may coexist with other diseases such as rheumatoid arthritis, scleroderma, 1335 dermatomyositis, Sjögren’s syndrome (sometimes with associated annular erythema), 1336 eosinophilic fasciitis, 1337 autoimmune thyroiditis, 1338 ulcerative colitis, 1339 myasthenia gravis, 1340 pemphigus, 1340 gout, 1341 alopecia areata, 1342 sarcoidosis, 1343 porphyria cutanea tarda,1344. and 1345. Sweet’s syndrome, 1346 psoriasis, 1347 pyodermatitis vegetans, 1348 cutaneous T-cell lymphoma, 1349 dermatitis herpetiformis, 1350 acanthosis nigricans, 1095 and various complement deficiencies.1351.1352.1353.1354. and 1355. Its relationship to Kikuchi’s disease (necrotizing histiocytic lymphadenopathy) remains a mystery. Patients with Kikuchi’s disease should be followed up long-term for the possible development of SLE.1356.1357. and 1358. Mycosis fungoides may masquerade as cutaneous lupus erythematosus. 1359 It has been suggested that some patients with photosensitive lupus erythematosus represent the coexistence of a photodermatosis such as polymorphic light eruption. 1360 Many of these associated conditions represent the chance coexistence of the two diseases, although the occurrence of SLE with diseases such as scleroderma, dermatomyositis, and rheumatoid arthritis has been included in the concept of mixed connective disease, an ill-defined condition with various overlap features and the presence of ribonucleoprotein antibody (see p. 310).1361. and 1362.
Joint symptoms, serositis and renal disease are frequent. 1095 Lymphocytopenia is common and correlates with the presence of autoantibodies targeting nuclear antigens.1363. and 1364. It is also a highly sensitive marker of systemic involvement but with low specificity. 1365 Rare manifestations include vegetations on the valve leaflets in the heart, vasculitis,1366. and 1367. diffuse pulmonary interstitial fibrosis, hemophagocytic syndrome, 1368 laryngeal lesions, 1369 mucosal involvement,1106. and 1370. peripheral neuropathy, and ocular involvement. 1095 Neurological manifestations are not uncommon and occasionally these are thromboembolic in nature, related to the presence of circulating anticardiolipin antibodies (the ‘lupus anticoagulant’) (see p. 201).1371.1372. and 1373. Cutaneous infarction1374.1375. and 1376. and ulceration1377 are other rare manifestations of this circulating antibody. However, digital necrosis can occur in patients with SLE in the absence of this antiphospholipid syndrome. 1378
SLE usually runs a chronic course with a series of remissions and exacerbations. The commonest causes of death are renal failure and vascular lesions of the central nervous system. 1379 Infection is another mode of death, usually related to immunosuppression. 1380 The 10-year survival rate currently exceeds 90%. 1095 In a recent review of 57 children with SLE, eight had died, six from severe infection and two from renal failure. 1381
Various laboratory investigations are undertaken in the diagnostic study of patients with suspected lupus erythematosus.1382.1383. and 1384. The LE cell preparation is now of historical interest only and it has been replaced as a screening test by the immunofluorescent detection of circulating antinuclear antibodies (ANA). 1385 The presence of LE bodies in vivo may have some diagnostic value. 1386 Various patterns of immunofluorescence, corresponding to different circulating antibodies, can be seen; the incidence of positivity depends on the substrate used. 1387 A homogeneous staining pattern is usually obtained. This test is positive in more than 90% of untreated patients, many of the negative cases belonging to the subset with anti-Ro/SSA antibodies (see p. 60). A recent study found that patients with SLE and positive ANA had a significantly higher frequency of renal disorders than those with negative ANA. 1388
Much more specific for the diagnosis is the detection of antibodies to double-stranded DNA. They are found in over 50% of cases and the titer may be used to monitor the progress of treatment. 1095 The presence of these antibodies is often associated with renal disease. They are usually detected by a radioimmunoassay method.
Other antibodies may also be detected, including rheumatoid factor and antibodies to extractable nuclear antigen (ENA) 1177 and Ro60. 1389 Anti-α-fodrin antibodies, commonly found in patients with Sjögren’s syndrome, are seen in a small number of patients with SLE. 1390 Antineutrophil cytoplasmic antibodies (ANCA) have been present in many patients with minocycline-induced lupus-like syndrome. 1391 They may also be present in other cases of systemic lupus erythematosus. 1392 False-positive serological tests for syphilis are sometimes present. 1095 Antibodies to cytoplasmic keratin proteins in the epidermis have been detected in patients with SLE. Their presence appears to correlate with the finding of cytoplasmic deposits of immunoglobulin in epidermal keratinocytes.1393. and 1394. Antibodies to basement membrane antigens are sometimes found in the sera of patients with no evidence of bullous lesions. 1395
Altered immunity, drugs, viruses, genetic predisposition, hormones and ultraviolet light may all contribute to the etiology and pathogenesis of lupus erythematosus. 1095 Immunological abnormalities are a key feature. Various autoantibodies are often present and high levels of antibodies against double-stranded DNA have been considered specific for SLE. Immune complexes are found in about 50% of affected individuals1172 and those containing DNA appear to be responsible for renal injury. Vascular injury may result from the deposition of these complexes. 1396 The link between the lupus band and the pathogenesis remains controversial, because immunoglobulins and complement components, including the membrane attack complex (MAC), can be found in both lesional and non-lesional skin of patients with SLE. 1397 CD59 (protectin) is expressed in non-lesional skin (and not in normal controls) in which complement activation has occurred. CD59 acts specifically to inhibit the terminal pathway of complement by blocking the formation of MAC. Although some MAC is found in non-lesional skin, as already stated, it seems that CD59 expression keeps this in check. 1397 There have been conflicting reports on the role of the various T-cell subsets.1398.1399. and 1400. However, CD4+ αβ T cells infiltrate the papillary dermis and appear to play some role in the basal damage. 1401 There is a slight predominance of CD4+ over CD8+ cells. Increased cytokine production, particularly IFN-γ, has been noted.1402. and 1403. A similar mechanism appears to be responsible in discoid lupus erythematosus. 1404 Infiltrating lymphocytes carrying CXCL10 in their granules might amplify the lesional inflammation and be responsible for the chronic course of this disease. 1403 It appears that dysregulation of T lymphocytes causes the activation of B cells, producing various autoantibodies of pathogenetic significance. 1405 Interestingly, a chimeric CD4-monoclonal antibody has been used successfully in the treatment of severe cutaneous lupus erythematosus. 1406 Other immunological findings include a reduction in epidermal Langerhans cells, loss of HLA-DR surface antigens on dermal capillaries, and a small percentage of Leu 8-positive cells in the dermal infiltrate.1399.1407. and 1408. HLA-DR is expressed on epidermal keratinocytes, while ICAM-1 is expressed on keratinocytes, dermal inflammatory cells, and endothelial cells. 1408
Matrix metalloproteinases, which contribute to tissue destruction, regeneration, inflammation and apoptosis, are abundantly expressed by keratinocytes in all major forms of cutaneous lupus erythematosus. 1409 In particular, MMP-3, -10, -19, and -26 are overexpressed while MMP-7 is detected in keratinocytes in regions of edema and vacuolization. 1409
In a small but important proportion of cases the onset of systemic lupus is quite clearly related to the ingestion of drugs. 1095 Those incriminated include procainamide, 1410 isoniazid, hydralazine, 1411 quinidine, 1412 minocycline,1413.1414.1415.1416. and 1417. penicillamine, 1418 sulfonamides, 1419 rifampicin, 1420 terbinafine,1421. and 1422. chlorpromazine, 1423 phenylbutazone, etanercept, 1424 hydrochlorothiazide, 1425 methimazole,1426. and 1427. carbamazepine,1428. and 1429. atenolol, 1430 practolol, phenobarbital, and phenytoin. A recent review also lists allopurinol, captopril, clonidine, danazol, ethosuximide, griseofulvin, lithium, lovastatin, mesalazine, methyldopa, penicillin, piroxicam, primidone, propylthiouracil, streptomycin, thiamazole, trimethadione, and valproate. 1384 A new anti-angiogenesis drug (COL-3) used in the treatment of cancer can also induce SLE. 1431 Oral contraceptives may sometimes result in a flare-up of the disease. Withdrawal of the drug is usually followed by slow resolution of the process. Exposure to insecticides has also been incriminated.1432. and 1433. Procainamide-induced SLE, which is the best studied of the drug-related cases, has a low incidence of renal involvement. 1095 High titers of leukocyte-specific ANA are present in those with clinical disease. 1434
A subset of drug-induced lupus erythematosus is characterized primarily by cutaneous disease and the usual presence of anti-Ro/SSA antibodies. The most common drugs associated with this disease are antihypertensive drugs, hydrochlorothiazide, calcium channel blockers, ACE inhibitors, griseofulvin, and terbinafine. 1435 A study published in 2007 concluded that long-term exposure to statins may be associated with drug-induced lupus erythematosus and other autoimmune disorders. 1436 Fatal cases have been reported despite early drug discontinuation and aggressive systemic immunosuppressive therapy. 1436 Antihistone antibodies are also found in drug-induced lupus.1437. and 1438.
The role of viruses is still controversial. Structures resembling paramyxovirus have been demonstrated on electron microscopy, particularly in endothelial cells, in SLE and also in discoid lupus erythematosus. 1439 There is doubt about the nature of these inclusions. Rare cases associated with parvovirus B19, and with HIV infection, have been reported.1214. and 1440. Epstein–Barr virus has also been incriminated. 1441
Familial cases have been recorded, usually in siblings, but also in successive generations. The disease has been observed in numerous pairs of identical twins. Several HLA types have been incriminated;1442. and 1443.one of these is found on chromosome 6p21. 1444 A recent study has shown an association between SLE and numerous genes, some with known immune-related functions. 1444 They include IRF5 on chromosome 7q32, ITGAM on 16p11.2, KIAA1542 on 11p15.5, and PXK on 3p14.3. 1444 A novel single-nucleotide polymorphism of the Fcγ receptor IIIa gene is associated with genetic susceptibility to SLE in Chinese populations. 1445
The role of sunlight in inducing and exacerbating cutaneous lupus of all types is well documented.1446.1447.1448. and 1449. Ultraviolet (UV)-B irradiation is the most frequent inducer of skin lesions in photosensitive lupus, although very high doses of UVA may trigger lesions in some patients.1199. and 1450. If an extended phototesting protocol is used, almost all patients with lupus erythematosus have evidence of aberrant photosensitivity. 1451 The mechanism of action appears to be the stimulation of keratinocytes to translocate cytoplasmic and nuclear antigens, such as SSA and SSB. Ultraviolet irradiation also stimulates keratinocytes and fibroblasts to release cytokines, such as tumor necrosis factor-α and interleukin-1α. A rare allele (-308A, TNF2) of the TNF-α promoter gene is strongly linked to subacute lupus erythematosus. 1450 Sunlight-induced damage of cellular DNA contributes ultimately to the formation of immune complexes which may be of pathogenetic significance.
To assist in the evaluation of clinical therapeutic trials, the Cutaneous Lupus Erythematosus Disease Area and Severity Index was developed. 1454 It is a useful tool for measuring clinical response. The treatment of SLE in which there is involvement of other organs is beyond the scope of this book. Cutaneous lesions are usually treated in the same way that skin lesions in the subacute form are managed. Photoprotection may also be required. 1451 There is one report of a patient with vasculitis in SLE who responded to rituximab, after failing to respond to mycophenolate mofetil, high-dose methylprednisolone, and intravenous immunoglobulin. 1367
The cutaneous lesions of systemic lupus erythematosus show prominent vacuolar change involving the basal layer. Civatte body formation is not usually a feature. Edema, small hemorrhages, and a mild infiltrate of inflammatory cells, principally lymphocytes, are present in the upper dermis. Eosinophils may be present in drug-induced cases and in urticarial lesions. Flame figures were present in one urticarial case. 1455 Fibrinoid material is deposited in the dermis around capillary blood vessels, on collagen and in the interstitium. It sometimes contributes to thickening of the basement membrane zone (Fig. 3.29). The main constituents of this thickened zone are type IV and type VII collagen. 1456 Mucin can be demonstrated by special stains1457 and its presence may be helpful in distinguishing the lesions of SLE from polymorphic light eruption. 1458 The term ‘cutaneous lupus mucinosis’ has been used for cases with abundant dermal mucin. 1323 A vasculitis, usually of leukocytoclastic type, is sometimes present. It may be complicated by thrombosis. Extravascular necrotizing palisaded granulomas, as originally described in Churg–Strauss syndrome, were present in one case. 1459
Acute lupus erythematosus. There is thickening of the basement membrane zone as well as vacuolar change and Civatte bodies. (H & E)
The subset of patients who have antibodies to Ro/SSA show additional vascular changes not usually seen in those without these antibodies. They include telangiectasia, endothelial cell necrosis and luminal deposits of fibrin. 1214
In follicular lupus erythematosus the interface changes are localized to the follicular infundibulum. 1322 Perivascular inflammation is present, allowing a distinction from lichen planopilaris.
In chilblain lupus, a lichenoid (vacuolar interface) reaction overlies a lymphocytic vasculitis involving both the superficial and deep plexuses.
In mucosal lupus, which affects predominantly the lips and buccal mucosa, a lichenoid mucositis with a band-like and deeper perivascular infiltrate of lymphocytes and some plasma cells is present. 1370 The cytokeratin profile is that of hyperproliferative epithelium with the expression of CK5/6 and CK14 in all epithelial layers, CK16 in the suprabasal layer and CK10 in the prickle cells. 1370
Neutrophils are sometimes present in the upper dermis in non-bullous cases. 1460 They are much less frequent than is seen in Sweet’s syndrome, except the very early stages of that disease. They are both perivascular and interstitial with leukocytoclasis. 1460 None of the patients progressed to bullous lupus erythematosus. Recently Pincus, McCalmont, and LeBoit have reported five patients with systemic lupus erythematosus and unusual neutrophilic infiltrates. 1461 There were some similarities in different cases to urticaria (but the infiltrate was too heavy), Sweet’s syndrome (but the infiltrate was confined to the papillary dermis and lacked edema), and palisaded neutrophilic and granulomatous disease (but the changes were in the papillary dermis). 1461 A similar finding has also been reported in neonatal lupus erythematosus. 1462
Hematoxyphile bodies – altered nuclei that are the tissue equivalent of the LE cells in the blood – are found rarely in the skin, in contrast to visceral lesions in which they are not infrequent.
The incidence of a positive lupus band test will depend on the site biopsied, 1463 and the duration of the lesion biopsied. Lesions should be of 2–3 months’ duration. Involved skin is positive in almost 100% of cases, while uninvolved skin from sun-exposed areas is positive in about 90% of cases. 1464 Biopsies of uninvolved skin from sun-protected areas are positive in only one-third of cases. Positive tests are obtained from sun-exposed skin in one-third or more of normal controls,1465.1466. and 1467. although the staining pattern is usually weak. 1465 There is a marked predominance of IgM ± C3. 1468 IgG3 is the predominant subclass of IgG deposited. 1469 The lupus band test colocalizes with collagen VII. 1470 Circulating basement membrane zone antibodies may participate in the formation of the lupus band. 1471 The lupus band test may be negative in remissions, early lesions, treated lesions, and some cases of drug-induced lupus erythematosus. 1472 It is also negative in UV-induced skin lesions, although their histopathological features are similar to those of primary lupus erythematosus of the different subtypes. 1199 The test is useful in excluding diseases that are clinically similar to SLE. 1473 Membrane attack complex (MAC) is deposited in a granular pattern along the basement membrane zone in approximately 75% of patients with cutaneous lupus erythematosus. 1474 It is also found in subacute lupus erythematosus. 1475 It may be a useful adjunct to the lupus band test. 1474 Another finding on direct immunofluorescence is epidermal nuclear staining, usually for IgG. It is found in only a small percentage of cases but it may correlate with oral involvement. 1476 Immunoelectron microscopy has shown that the immunoglobulin deposits are predominantly in the papillary dermis, just beneath the basal lamina. 1477 The deposition appears to damage both type IV and type VII collagen. 1478 DNA is a major component of the complexes.
The traditional variants of lupus erythematosus (discoid, subacute, and systemic) have already been discussed. This section covers uncommon, but distinct, clinicopathological variants.
Neonatal lupus erythematosus is a rare syndrome1479.1480.1481.1482. and 1483. characterized by a transient lupus dermatitis developing in the neonatal period, accompanied by a variety of hematological and systemic abnormalities,1484. and 1485. including congenital heart block.1486. and 1487. Congenital presentation is rare. 1488 Cutaneous lesions resemble those seen in subacute cutaneous lupus erythematosus; 1489 telangiectatic macules may be a feature.1490. and 1491. Periorbital, scalp, and extremity lesions are common. 1492 Papules on the plantar surface of both feet are a rare presentation. 1493 So too is a presentation mimicking Langerhans cell histiocytosis. 1494 Depigmented lesions are rare; 1495 so too are cutaneous erosions1496 and lupus panniculitis. 1497 Approximately 20% of the mothers have SLE at the time of the birth and a similar percentage will subsequently develop it. 1486 Various factors have been proposed to explain clinical disease in the fetus in the absence of disease in the mother. 1498 Other mothers may have Sjögren’s syndrome or, uncommonly, a vasculitis. 1499 The Ro/SSA antibody is present in infants and mothers in nearly all cases1489.1492.1500.1501.1502. and 1503. and it has been suggested that this is of maternal origin and crosses the placenta, where it is subsequently destroyed by the infant.1504. and 1505. Antibodies to α-fodrin, 1506 La/SSB and U1RNP have been detected in some cases.1507.1508.1509.1510. and 1511. The anticardiolipin antibody has been reported in a baby with neonatal lupus erythematosus. 1512 Successive siblings may be affected with this condition.1513. and 1514. Persistent scarring, atrophy, and depigmentation may result. 1515
The histological features resemble those seen in subacute lupus erythematosus. 1516
A rare form of SLE, bullous lupus erythematosus, is a skin eruption which clinically and histologically closely resembles dermatitis herpetiformis (Fig. 3.30).1517.1518.1519. and 1520. Rare clinical variants include a localized linear form1521 and one in which milia develop. 1522 The blisters are subepidermal with neutrophils in the papillary dermis and some lymphocytes around vessels in the superficial plexus. 1523 Bullae have been reported in a case of discoid lupus erythematosus1524 and also following steroid withdrawal in SLE. 1321 The antiphospholipid syndrome is a rare association. 1525 Linear or mixed linear and granular deposits of IgG are found along the basement membrane zone. 1526 IgA and/or IgM may also be present.1527.1528. and 1529. The immunoreactants are deposited beneath the lamina densa, 1523 and they are accordingly on the dermal side of salt-split skin. 1530 Electron microscopy confirms that the split is below the lamina densa. 1531
Bullous lupus erythematosus. There are neutrophils in the papillary dermis in this field. The blister is not shown. (H & E)
There appear to be at least two immunologically distinct subtypes characterized by the presence or absence of circulating and/or tissue-bound autoantibodies to type VII collagen.1528.1532.1533. and 1534. Patients with autoantibodies to type VII collagen are similar but not identical to patients with epidermolysis bullosa acquisita1535 (see p. 147). The autoantibodies appear to recognize the same non-collagenous (NC1) domain. 1536 In other cases, various components of the basement membrane zone may be targeted. In one case there were autoantibodies to the bullous pemphigoid antigen (BP230, BPAg1), laminin-5, laminin-6, and type VII collagen. 1537 In another case the autoantibodies were to BPAg1 alone. 1538
Bullous systemic lupus erythematosus is treated with dapsone, corticosteroids and other immunosuppressive agents. The response to dapsone is often dramatic and supports its use as first-line therapy for this condition. 1531 Antimalarials can also be used, but patients with concurrent porphyria cutanea tarda may exhibit a toxic response. 1201
Lupus panniculitis (lupus profundus) presents clinically as firm subcutaneous inflammatory nodules, from 1–4 cm or more in diameter, situated on the head, neck, arms, abdominal wall, thighs, or buttocks.1539. and 1540. There is a greater frequency of periorbital edema as the initial manifestation of lupus profundus in black South Africans compared to other published series. 1541 There are isolated reports of breast involvement (lupus mastitis),1542.1543. and 1544. and also of linear lesions. 1545 Rare cases have been reported in childhood. 1120 Lupus panniculitis is a rare complication which may precede the development of overt systemic or discoid lupus erythematosus. 1546 A patient with widespread lesions in the setting of a partial deficiency of C4 has been reported. 1547 In some cases the lesions subside without any other sign of the disease. 1548 The clinical diagnosis may be difficult if there are no other manifestations of lupus erythematosus. The lesions may be misdiagnosed as deep morphea. 1549 Lupus panniculitis is discussed in more detail with the panniculitides (Ch. 17, p. 468). Lupus panniculitis may be responsive to antimalarials. 1201
Dermatomyositis is characterized by the coexistence of a non-suppurative myositis (polymyositis) and inflammatory changes in the skin.1550.1551.1552.1553. and 1554. Cutaneous lesions may precede the development of muscle involvement by up to 2 years or more.1555.1556. and 1557. Cases without muscle involvement (amyopathic dermatomyositis, dermatomyositis sine myositis) exist.1558.1559.1560.1561.1562.1563. and 1564. Amyopathic dermatomyositis is defined as the finding of dermatomyositis in the absence of any clinical or laboratory signs of muscle disease for at least 6 months (formerly 2 years) after the onset of skin pathology.1565. and 1566. It accounts for 10–20% of the total population of dermatomyositis patients seen in referral clinics in the United States. 1566 There is a strong association between amyopathic dermatomyositis and nasopharyngeal carcinoma in China. 1567 Its association with familial polyposis coli was probably fortuitous. 1568 An ‘adermatopathic’ variant of dermatomyositis has also been postulated. 1566
Dermatomyositis may occur in either sex and at any age. Those cases commencing in childhood are sometimes considered as a separate clinical group (juvenile dermatomyositis), because of the greater incidence in them of multiorgan disease.1569.1570.1571.1572. and 1573. Rarely this includes a necrotizing vasculitis that may involve the gut and other organs with a fatal outcome. 1569 The initial physical and laboratory findings in patients with juvenile dermatomyositis may be non-specific. Instead of the heliotrope rash and Gottron’s papules classically associated with dermatomyositis (see below), there may be a non-specific extremity rash, periungual erythema, and sometimes pruritus. 1574 An amyopathic variant also occurs. 1575 Vasculopathy does not seem to occur in this amyopathic form of juvenile dermatomyositis. 1575 Other manifestations of juvenile dermatomyositis include lipoatrophy, generalized hypertrichosis, and infrapatellar hypertrichosis1576 Associations include the 22q11.2 deletion syndrome. 1577
The skin lesions in adult dermatomyositis are violaceous or erythematous, slightly scaly lesions with a predisposition for the face, shoulders, the extensor surfaces of the forearms and the thighs. 1578 Poikilodermatous features (telangiectasia, hyperpigmentation, and hypopigmentation) may be seen. 1579 Photosensitivity is sometimes present. 1580 Pruritus is not uncommon, but it has not been highlighted in the literature. 1581 Other characteristic findings are nail fold changes, gingival telangiectases, 1582 purplish discoloration and edema of the periorbital tissues (heliotrope rash), and atrophic papules or plaques over the knuckles (Gottron’s papules). 1579 Plaques of calcification sometimes develop.1583. and 1584. Unusual presentations include severe periorbital edema, 1585 an isolated flagellate eruption on the back,1586.1587. and 1588. a plaque-like mucinosis,1589. and 1590. the presence of follicular keratotic papules with some features of pityriasis rubra pilaris,1591.1592.1593. and 1594. lesions resembling malignant atrophic papulosis, 1595 a panniculitis,1596. and 1597. stasis dermatitis, 1598 erythroderma,1599. and 1600. or vesiculobullous eruption. 1601 A patient with vesiculobullous disease and a panniculitis but without muscle disease has been reported. 1602 In another patient with acute onset vesiculobullous dermatomyositis, massive mucosal necrosis of the intestines occurred. 1603 A Dermatomyositis Skin Severity Index has recently been developed and validated. 1604 This will allow a comparison of various treatment modalities in future clinical trials.
Other clinical features include the presence of proximal muscle weakness and elevation of certain serum enzymes such as creatine phosphokinase.1578. and 1605. Muscle ultrasound is sometimes abnormal in patients with dermatomyositis and normal muscle enzyme levels. 1606 Raynaud’s phenomenon, dysphagia, Sjögren’s syndrome, morphea profunda, 1607 retinopathy, and overlap features with scleroderma1608. and 1609. and with lupus erythematosus sometimes occur.1579. and 1610. Interstitial lung disease is an uncommon but debilitating complication which is usually associated with the presence of the anti-Jo-1 antibody.1611.1612.1613.1614. and 1615. Digital infarcts showing microangiopathy may occur in patients with severe pulmonary disease. 1616 Other autoantibodies, such as to histone and Ro/SS-A, are sometimes present.1617. and 1618.
An underlying malignancy is present in 10% or more of cases.1619.1620.1621.1622.1623.1624.1625.1626.1627.1628. and 1629. In one series the prevalence of malignancy was high (23%). 1588 Predictive factors for malignancy include male gender and older age of onset.1630. and 1631. Although the malignancies have spanned nearly every organ in the body, specific variants published in recent years have included nasopharyngeal carcinoma,1632. and 1633. ovarian cancers,1624.1625. and 1634. melanoma, 1635 breast cancer, 1633 squamous cell carcinoma of the penis, 1636 transitional cell carcinoma of the bladder,1627. and 1637. lymphoma, 1638 myeloma, 1629 and cancers of the esophagus, 1639 lung, stomach, and colon. 1640 The cutaneous manifestations may precede the diagnosis of the malignancy by up to a year or more. Amyopathic cases of paraneoplastic dermatomyositis have been reported. 1641 Dermatomyositis occurring in patients with stage IV melanoma has a poor prognosis.1642. and 1643. Subepidermal vesiculation is an uncommon finding in dermatomyositis; its presence may be related to the occurrence of an internal malignancy.1601. and 1644. Bullous pemphigoid has also been recorded as a coexisting disease. 1645
A dermatomyositis-like syndrome has been associated with certain viral illnesses, including parvovirus B19, 1646 toxoplasmosis, 1647 and leishmaniasis. 1648 It has followed administration of hydroxyurea,1649.1650.1651.1652.1653.1654.1655.1656.1657.1658. and 1659. omeprazole, 1660 carbimazole, 1661 terbinafine, 1662 tegafur, interferon-α, BCG vaccination, the various statins, 1662 cyclophosphamide, 1663 etoposide, 1663 a herbal medicine, 1664 and penicillamine. 1578 A heliotrope-like eruption mimicking dermatomyositis occurred in a patient receiving imatinib mesylate. 1665 Dermatomyositis, including the amyopathic form, has developed in pregnancy with rapid improvement following delivery1666. and 1667. and in the postpartum period. 1668 It has been reported in a patient with hereditary complement (C9) deficiency, 1669 and in a patient with chronic graft-versus-host disease. 1670 Cytoplasmic inclusions resembling the paramyxovirus-like structures seen in lupus erythematosus (see p. 62) have also been found in blood vessels in cases of dermatomyositis. 1671
The etiology and pathogenesis are unknown but immunological mechanisms are certainly involved. In addition to the various autoantibodies that are often present, activated T cells and natural killer cells have been demonstrated in biopsied muscle. 1578 In the skin, the infiltrate consists predominantly of macrophages expressing HLA-DR and of T cells, especially of the CD4 subset. 1672 Plasmacytoid dendritic cells (bone-marrow derived dendritic cells with an ability to secrete large amounts of IFN-α in vivo after appropriate stimulation, including viral infection) are present in the skin. 1673 These cells, which stain with CD123, play a central role in the pathogenesis of lupus erythematosus, 1673 but in contrast to lupus erythematosus where they are found in the dermis, in dermatomyositis there is a preferential epidermal localization. 1673 The deposition of membrane attack complex of complement (MAC) has been demonstrated along the dermoepidermal junction and in some dermal blood vessels. 1674 Endothelial cell injury may play an important role in producing some of its clinical manifestations, including lung disease. 1675 There are elevated levels of soluble vascular cell adhesion molecule-1 (sVCAM-1). 1676 There is increasing evidence that a type I interferon-driven immune response, and the recruitment of potentially autoreactive T cells via IP10/CXCR3 interaction are involved in the pathogenesis of dermatomyositis skin lesions.1677.1678. and 1679. Plasmacytoid dendritic cells appear to be an important source of these type I interferons. 1679
Spontaneous remission without therapy is rare in dermatomyositis. 1680 Although there are no double-blind randomized trials, oral corticosteroids are generally considered the primary therapy for dermatomyositis with muscle disease.1680. and 1681. They are not effective in all patients and they are frequently associated with adverse effects. Antimalarials, methotrexate, azathioprine, chlorambucil, cyclosporine, and cyclophosphamide have been used in combination with low-dose corticosteroids, or as an alternative treatment, with varying degrees of success. 1682 Cutaneous reactions to hydroxychloroquine occur in approximately 30% of patients. 1683 Mycophenolate mofetil may also be used as an effective corticosteroid-sparing agent for some patients. 1682 Leflunomide has been trialed as adjuvant therapy. 1684 Intravenous immunoglobulin has also been used.
Therapy using anti-TNF-α drugs have been tried with variable results. Tamoxifen, which also has anti-TNF-α effect, has been successful in a few cases. 1685 Rituximab, a monoclonal anti-CD20 antibody, has also been used. 1686 Its effect on muscle disease may be better than on skin disease. 1687
The treatment of amyopathic juvenile dermatomyositis is controversial in the absence of appropriate controlled trials. Some adopt a conservative approach while others treat aggressively with immunosuppressive agents (prednisone, methotrexate) in an attempt to minimize the risk of progression to myositis. 1575 One study showed that aggressive treatment resulted in improved outcome and a decreased incidence of calcinosis. 1688
The histological changes are quite variable. At times the changes are subtle (Fig. 3.31) with only a sparse superficial perivascular infiltrate of lymphocytes, associated with variable edema and mucinous change in the upper dermis. 1689 More often, there are the features of a lichenoid tissue reaction consisting of vacuolar change in the basal layer. Only occasional apoptotic keratinocytes are present, if any (Fig. 3.32). A few neutrophils are sometimes present in the infiltrate, particularly in those cases with fibrinoid material in the papillary dermis and around superficial vessels. Diffuse dermal neutrophilia and leukocytoclastic vasculitis are rare findings.1690. and 1691. The basement membrane is often thickened. These appearances are indistinguishable from those of acute lupus erythematosus.
Dermatomyositis. The changes are quite subtle with mild basal vacuolar change and several colloid bodies in the papillary dermis. The appearances may be indistinguishable from acute lupus erythematosus although in the latter disease basement membrane thickening may be more pronounced and colloid bodies less frequent than in dermatomyositis. (H & E)
Dermatomyositis. The lichenoid reaction pattern is more obvious in this case. The appearances are indistinguishable from cutaneous lupus erythematosus. (H & E)
At other times, there are additional features of epidermal atrophy, melanin incontinence, and dilatation of superficial vessels (poikilodermatous changes). 1689 A mild sclerodermoid tissue reaction of variable depth is sometimes present. This may be a consequence of microvascular injury. Lung disease is usually present in these cases. 1675 A biopsy from a Gottron’s papule will show mild hyperkeratosis and some acanthosis, in addition to the basal vacuolar change. 1692 Mucin deposition is present in nearly 40% of cases. The changes are similar to those observed in dermatomyositis at other cutaneous sites. 1693
Unusual findings include subepidermal vesiculation, a lobular panniculitis, and dystrophic calcification.1596.1597.1694.1695. and 1696. The vesicular lesions are cell poor with abundant edema fluid. Necrosis of the roof sometimes occurs. An osteogenic sarcoma developed in an area of heterotopic ossification in one case of dermatomyositis. 1697
In the Wong type of dermatomyositis, in which pityriasis rubra pilaris-like lesions are present, there is follicular hyperkeratosis with destruction of hair follicles. Cornoid lamellation has been seen in one of these cases. 1593
Intercellular deposits of immunoglobulins have been reported in the epidermis of nail fold biopsies. 1698 Colloid bodies containing IgM are sometimes quite prominent in the papillary dermis. The lupus band test is usually negative although it was positive in a significant number of cases in one series. 1699 Vascular deposits of C5b–9 are found in dermatomyositis but not in lupus erythematosus. 1700 Immunoglobulins, including IgA, have been reported in muscle biopsies in dermatomyositis. 1701
The poikilodermas are a heterogeneous group of dermatoses characterized clinically by erythema, mottled pigmentation, and, later, epidermal atrophy. These changes result from basal vacuolar change with consequent melanin incontinence and variable telangiectasia of blood vessels in the superficial dermis (Fig. 3.33). For this reason, Pinkus included the poikilodermatous pattern as a subgroup of the lichenoid tissue reaction. 1
The poikilodermatous reaction pattern in a case of mycosis fungoides. (H & E)
Four distinct groups of poikilodermatous dermatoses are found:
2. a stage in the evolution of early mycosis fungoides;
3. a variant of dermatomyositis and less frequently of SLE;
4. a miscellaneous group which may follow radiation, cold and heat injury, prolonged exposure to sunlight (poikiloderma of Civatte), and ingestion of drugs (arsenicals and busulfan) or which may occur in the evolution of chronic graft-versus-host reaction. 1702
The three genodermatoses mentioned above are distinct clinical entities, but the poikiloderma which may precede mycosis fungoides (poikiloderma atrophicans vasculare) is now regarded as an early stage in the evolution of mycosis fungoides. 1703 Likewise, poikilodermatomyositis represents a clinicopathological variant of dermatomyositis rather than a disease sui generis. Poikiloderma of Civatte is a controversial entity. It will be considered briefly below.1704. and 1705.
Over 250 cases of poikiloderma congenitale (Rothmund–Thomson syndrome; OMIM 268400) have now been reported in the English literature.1706. and 1707. It is an autosomal recessive, genomic instability syndrome. It is a multisystem disorder which affects principally the skin, eyes, and skeletal system.1708.1709.1710.1711.1712.1713. and 1714. There is a predisposition to malignancy. 1715 A reticular erythematous eruption commences in the first year of life and this is followed by the development of areas of hypo/hyperpigmentation. Warty keratoses may appear on the hands, elbows, knees, and feet. Other clinical features include a short stature, cataracts, hypogonadism, mental retardation, photosensitivity to ultraviolet A radiation,1706. and 1716. and, rarely, the development of skin cancers,1717. and 1718. including melanoma, 1719 hematological malignancies, 1720 including myelodysplasia,1721.1722. and 1723. and osteogenic sarcoma.1724. and 1725. Sometimes only a few of these features are present in an individual case, illustrating the variable presentation of this syndrome. 1726 Late onset has also been recorded. 1727 Reduced DNA repair capacity might be related to the photosensitivity in early childhood. 1728 There appears to be instability in chromosome 8, sometimes leading to trisomy 8 or other abnormalities. 1726 The condition appears to be genetically heterogeneous. Mutations in the RECQL4 helicase gene which maps to chromosome 8q24.31729 are present in about 60% of cases. 1719 This gene encodes a RecQ DNA helicase. 1715 It appears to have a role in the initiation of DNA replication and in sister chromatid adhesion. 1715 The RECQL4 gene has also been associated with two other diseases, the Baller–Gerold syndrome (OMIM 218600) and RAPADILINO syndrome (RAdial hypoplasia/aplasia, PAtellar hypoplasia/aplasia, cleft or highly arched PAlate, DIarrhea, DIslocated joints, LIttle size, LImb malformations, and slender NOse and NOrmal intelligence; OMIM 266280). They appear to be allelic with different phenotypic expressions. Poikilodermatous features are usually absent in this latter variant, 1730 but they have been recorded in the Baller–Gerold syndrome, in which craniosynostosis, radial ray defects, and growth retardation are present.
There are the usual poikilodermatous features of hyperkeratosis, epidermal atrophy, basal vacuolar change, rare apoptotic keratinocytes in the basal layer, numerous telangiectatic vessels, scattered dermal melanophages, and a variable upper dermal inflammatory cell infiltrate. 1727
The keratotic (warty) lesions show hyperkeratosis, a normal or thickened epidermis and some loss of cell polarity, with dyskeratotic cells also present. 1708
Hereditary sclerosing poikiloderma (of Weary) – OMIM 173700 – is an autosomal dominant disorder with many similar clinical features. 1731 However, there are linear hyperkeratotic and sclerotic bands in the flexural areas, sclerosis of the palms and soles, and clubbing of the nails. 1732 There have been no recent publications on this entity.
Kindler’s syndrome (OMIM 173650) is a rare autosomal recessive genodermatosis characterized by acral trauma-induced blistering that improves with age and by progressive poikiloderma in later life. 1733 There are variable degrees of photosensitivity beginning in childhood. 1734 Other clinical features include webbing of the fingers and toes, nail dystrophy, pitted palmoplantar keratoderma, 1735 periodontal disease, phimosis, 1736 esophageal and anal strictures, and gastrointestinal erosions.1733.1737.1738.1739. and 1740. Clinical heterogeneity is well recognized.1741. and 1742. Kindler’s syndrome is included as a category of epidermolysis bullosa in the new classification of this disease (see Ch. 6, p. 141).
Kindler’s syndrome is due to a loss of function mutation in the KIND1 gene, also known as C20orf42. 1740 The gene encodes a protein called kindlin-1 which is involved in linking the actin cytoskeleton to integrin-associated platforms in the extracellular matrix.1743. and 1744. It is mainly found in keratinocytes. Loss of kindlin-1 leads to decreased cell adhesion, reduced proliferation of keratinocytes, and increased apoptosis. 1741 Kindler’s syndrome is the first inherited disorder that has been found to result from primary defects in the actin cytoskeleton/focal contacts. 1741 More than 25 different mutations have so far been described in KIND1, but they do not provide an explanation for the clinical heterogeneity of this disease.1741.1743.1745. and 1746. Furthermore, KIND1 gene expression and kindlin-1 protein labeling are not always reduced in this syndrome. 1747
Some cases are initially misdiagnosed as dystrophic epidermolysis bullosa, but there is no mutation in COL7A1.1748. and 1749. Kindler’s syndrome was initially thought to represent the association of poikiloderma congenitale with dystrophic epidermolysis bullosa.1750.1751.1752. and 1753. Similar cases have been reported under the title hereditary acrokeratotic poikiloderma.1754. and 1755.
There is mild hyperkeratosis, epidermal atrophy, and basal vacuolar change. Rare apoptotic keratinocytes are present in the lower epidermis. There is some telangiectasia and scattered dermal melanophages. There is usually only a sparse superficial dermal infiltrate of lymphocytes.
The bullae in Kindler’s syndrome are subepidermal and cell poor, although ultrastructural studies have shown that the split can occur at several levels within the dermoepidermal junction zone, including within the basal cells.1734.1756.1757. and 1758. There is also extensive reduplication of the lamina densa.1736.1759. and 1760.
Congenital telangiectatic erythema (OMIM 210900) is a rare autosomal recessive disorder usually known by the eponymous designation Bloom’s syndrome. In addition to the telangiectatic, sun-sensitive facial rash, there is stunted growth, proneness to respiratory and gastrointestinal infections, chromosomal abnormalities, and a variety of congenital malformations.1761.1762. and 1763. The facial rash has lupus-like qualities. 1764 Various chromosomal breakages are found in cultured lymphocytes, the most characteristic being a high rate of sister chromatid exchanges during metaphase. 1765 As a consequence, there is a significant tendency to develop various malignancies, 1766 particularly acute leukemia and lymphoma.1761. and 1767. The gene responsible, RECQ3, has been mapped to chromosome 15q26.1. 1763 Other RECQ helicase defects are found in Werner’s syndrome (a defect in RECQ2), and Rothmund–Thomson syndrome (RECQL4).
The facial rash consistently shows dilatation of dermal capillaries. There is usually only a mild perivascular infiltrate of lymphocytes. Basal vacuolar change may occur but does not usually result in pigment incontinence.
Dyskeratosis congenita is a rare, sometimes fatal genodermatosis characterized primarily by the triad of reticulate hyperpigmentation, nail dystrophy, and leukokeratosis of mucous membranes.1768.1769. and 1770. Other less constant features include a Fanconi-type pancytopenia,1771.1772. and 1773. eye and dental changes, mental deficiency, deafness, 1774 intracranial calcification, 1775 palmoplantar hyperkeratosis, scarring alopecia, 1776 esophageal and anal strictures, 1777 choanal atresia, 1778 Chiari 1 malformation, 1779 and an increased incidence of malignancy, particularly related to the mucous membranes,1774.1780. and 1781. and, less commonly, the skin. 1782 Bone marrow failure is the major cause of premature death.
Although found predominantly in Caucasian males, it has been reported in several races and occasionally in females. 1783 Most cases are inherited as a sex-linked recessive trait, 1774 but kindreds with both autosomal dominant and autosomal recessive inheritance have been reported.1784. and 1785. The various types of dyskeratosis congenita are listed in Table 3.6. In all characterized cases of dyskeratosis congenita, the causative mutations are present in components of the telomerase complex. Chromosomes shorten during DNA replication and it is the function of telomerase to add telomere repeats (a repeat comprises six nucleotides) to the ends of chromosomes. 1785 The gene for X-linked recessive dyskeratosis congenita, DKC1, which encodes a 514 amino acid protein, dyskerin, is located at Xp28. 1786 The disease is predominantly caused by missense mutations in this gene. 1787 It is thought that dyskerin is a nucleolar protein that is responsible for some early steps in ribosomal-RNA processing. 1786 This defect appears to be associated with a more severe phenotype than the autosomal dominant form, which has heterozygous mutations in either TERC or TERT, the RNA and enzymatic components of telomerase, respectively. 1785 The majority of documented cases of this form involve TERC mutations. The TINF2 gene appears to be another candidate gene. Autosomal recessive dyskeratosis congenita is more enigmatic. Recently, a homozygous mutation in NOP10 (NOLA3) was found in a consanguineous family. This mutation results in short telomeres and low TERC levels. 1785
The skin changes, which may resemble poikiloderma, usually develop on the face, neck, and upper trunk in childhood. It has been suggested that there may be pathogenetic features in common with graft-versus-host disease. 1768
Treatment with an anabolic steroid and hematopoietic growth factors can produce an improvement in hematopoietic function for some time, 1788 but eventually allogeneic stem cell transplantation is needed. Because of the pulmonary vascular complication, this procedure has not been particularly successful. 1785 In the future, gene therapy with the introduction of the wild-type form of the defective gene into stem cells should correct downstream defects in this disease. 1785
Usually there are mild hyperkeratosis, epidermal atrophy, prominent telangiectasia of superficial vessels, and numerous melanophages in the papillary dermis. Less constant features include mild basal vacuolar change, fibrosis of the upper dermis, and a mild lymphocytic infiltrate beneath the epidermis. 1768 Civatte bodies have not been recorded.
Poikiloderma of Civatte is a common dermatosis, particularly in Greece and other parts of Europe; it can produce cosmetic disfigurement. 1789 It is a neglected and controversial entity that has received little attention in other parts of the world. It most often affects fair-skinned individuals in their fourth to seventh decades. It is characterized by red to brownish, reticular patches with irregular borders and symmetrical distribution. It may involve the V and sides of the neck, the upper chest, and parts of the face. 1790 Erythemato-telangiectatic and pigmented types occur. The etiology is unknown but it is considered to be the cumulative effect of sun exposure exacerbated by the application of fragrances to the neck in combination with genetic predisposition and lighter skin phenotypes.1789. and 1791.
Sun protection and the avoidance of documented allergens should be practiced. 1789 Several patients who have been treated with pulsed dye laser have developed severe depigmentation. 1791 Depigmenting agents can be used as adjuvants in the pigmented variant of the disease. 1790
There is variable telangiectasia and melanin incontinence. Mild epidermal atrophy is sometimes present, particularly in older lesions. Some solar elastosis is invariably seen. Mild vacuolar change is sometimes present. There are sparse perivascular lymphocytes.
In addition to the dermatoses discussed above, a number of other important diseases may show features of the lichenoid reaction pattern. They have been discussed more fully in other chapters, but they are also included here for completeness. Only the salient histological features are mentioned.
In early lesions, the inflammatory infiltrate is quite heavy with band-like qualities mimicking lichen planus. Both vacuolar change and apoptotic basal keratinocytes are present. The infiltrate is eventually pushed downwards by an expanding zone of edema and sclerosis (see p. 313).
In the acute form of pityriasis lichenoides, pityriasis lichenoides et varioliformis acuta (PLEVA), there is a heavy lymphocytic infiltrate which obscures the dermoepidermal interface in much the same way as it does in erythema multiforme and fixed drug eruption (Fig. 3.34). This may be associated with focal epidermal cell death and overlying parakeratosis or confluent epidermal necrosis. 1792 The dermal infiltrate varies from a mild lymphocytic vasculitis to a heavy infiltrate which also extends between the vessels and is accompanied by variable hemorrhage. The dermal infiltrate is often wedge-shaped in distribution with the apex towards the deep dermis. PLEVA is considered further with the lymphocytic vasculitides in Chapter 8 (p. 230).
Pityriasis lichenoides (acute form). The dermoepidermal interface is obscured by the inflammatory cell infiltrate. (H & E)
There is increasing recognition that viral and putative viral infections may be followed by a spectrum of cutaneous reactions that often includes the lichenoid tissue reaction. Examples include lichenoid lymphocytic vasculitis (see p. 226) which occurs with persistent herpes simplex infection, the Gianotti–Crosti syndrome (see p. 116) 1793 and reactions resembling mild pityriasis lichenoides. A chronic lichenoid dermatosis has been reported in several patients as an unusual manifestation of both herpes simplex and varicella-zoster infection. There was a lichenoid reaction but no cytolytic host response. 1794 Late stages of pityriasis rosea (included here as a putative viral infection) often show prominent epidermal cell death. Basal vacuolar change is usually present in these various reactions.
Asymmetric periflexural exanthem of childhood, also known as unilateral laterothoracic exanthem, is a putative viral disease with pruritic, unilateral macules and papules. It has a distinctive perisudoral CD8+ infiltrate at the interface. Lymphocytes have also been found around the eccrine coils. Apoptotic basal keratinocytes are also present.1795. and 1796.
In some cases of perniosis, a mild lichenoid reaction is present. It is mostly focal. The changes are more prominent in cases of chilblain lupus (lupus pernio). There is usually no parakeratosis, unlike pityriasis lichenoides which also combines a lichenoid and vasculitic tissue reaction. In perniosis there is usually a thick layer of orthokeratin reflecting the acral site.
Paraneoplastic pemphigus (see p. 151) resembles erythema multiforme, with a lichenoid tissue reaction and dyskeratotic cells at different levels of the epidermis. Usually, foci of suprabasal acantholysis and clefting are also present. Subepidermal clefting has also been reported. A lichenoid variant of paraneoplastic pemphigus has been described without detectable autoantibodies. 1797
Some lesions of pigmented purpuric dermatosis may show lichenoid as well as purpuric and chronic vasculitic features (see p. 233). The presence of purpura and hemosiderin are important clues to the diagnosis.
A lichenoid contact dermatitis has been seen after contact with rubber and certain clothing dyes, and following contact with chemicals used in the wine industry. In two personally studied cases, there was a patchy, band-like dermal infiltrate of lymphocytes with a few eosinophils and very mild basal spongiosis.
A unique pattern of dyskeratosis has been reported in cases of adult-onset Still’s disease (fever, polyarthralgia, lymphadenopathy, evanescent rash).1798. and 1799. There are multiple dyskeratotic cells, singly or in aggregates, mainly located in the upper epidermis, including the stratum corneum. 1798 There are no associated lymphocytes. The presence of neutrophils in the dermal infiltrate is another characteristic feature. 1798
Some lesions of late secondary syphilis show a lichenoid reaction pattern (see p. 575). There is usually extension of the inflammatory infiltrate into the mid and deep dermis. Plasma cells are usually present in the infiltrate.
In lesions of porokeratosis, particularly the disseminated superficial actinic form, a lichenoid tissue reaction associated with a heavy superficial lymphocytic infiltrate can occur. A careful search will reveal the diagnostic cornoid lamella at the periphery of the infiltrate. The lichenoid infiltrate may be directed against the abnormal epidermal clones which emerge in this condition. Porokeratosis is considered in detail in Chapter 9 (p. 262).
The lichenoid reaction pattern is a prominent feature in lichenoid and fixed drug eruptions. In many other drug-induced cutaneous reactions, a very occasional Civatte body (apoptotic keratinocyte) may be seen in the basal layer or at a higher level within the epidermis. There may be an associated exocytosis of a few lymphocytes. Apoptotic cells are a valuable clue to the drug etiology of an otherwise non-specific spongiotic tissue reaction (see p. 116). These cells are usually easier to find in morbilliform drug eruptions.
In a phototoxic dermatitis there are scattered apoptotic keratinocytes (dyskeratotic cells, sunburn cells) at all levels of the epidermis. In severe cases confluent necrosis may be present. There is some telangiectasia of superficial dermal vessels, but very little dermal inflammation.
A patchy lichenoid reaction with associated melanin incontinence is seen in this uncommon condition (see p. 296).
A lichenoid reaction is present in some patients with erythroderma (see p. 507). Many of these cases may be drug induced.
A subset of patients with mycosis fungoides has lichenoid changes on biopsy. One study has found that lichenoid changes tend to be associated with intense pruritus and may connote a poor prognosis. 1800 The presence of basal epidermotropism, nuclear atypia in the lymphocytes, and the presence of eosinophils and sometimes plasma cells in the dermal infiltrate are helpful in identifying the underlying mycosis fungoides. 1800
The regression of viral warts, particularly plane warts, is associated with a lichenoid reaction pattern and exocytosis of cells into the epidermis. Keratinocytes in the stratum malpighii, presumably expressing viral antigen, are attacked by lymphocytes, resulting in death of the keratinocytes by apoptosis. Sometimes two or more lymphocytes ‘surround’ a keratinocyte, similar to the ‘satellite cell necrosis’ (lymphocyte-associated apoptosis) of graft-versus-host disease.
A lichenoid reaction pattern can be associated with a variety of epidermal tumors, where it appears to represent the attempted immunological regression of those lesions. This may be seen in seborrheic keratoses (the so-called ‘irritated’ seborrheic keratosis), solar keratoses (lichenoid solar keratoses) and intraepidermal carcinomas. The lichen planus-like keratosis represents a similar reaction in a solar lentigo and probably some other epithelial lesions.
A similar mechanism is involved in the partial regression of basal and squamous cell carcinomas and other cutaneous tumors. However, these circumstances do not conform to the definition of the lichenoid reaction pattern, namely basal epidermal cell damage. Accordingly they will not be considered further in this section.
In lichen amyloidosus there is an accumulation of filamentous material in basal cells, with their eventual death. The filamentous material is extruded into the dermis in a manner similar to the formation of colloid bodies. The basal cells possibly die by apoptosis but the accumulation of the filamentous material obscures this basic process (see p. 380).
In active lesions of vitiligo, careful search will often reveal an occasional lymphocyte in contact with a melanocyte. The destruction of melanocytes by lymphocyte-mediated apoptosis would explain the features of vitiligo (see p. 283).
A lichenoid reaction, localized to the red areas, is a rare complication in a tattoo.
A lichenoid reaction may be seen in several other circumstances in which it is not usually a feature. Examples include candidiasis of the lip and pityriasis rubra pilaris. A pin-point lichenoid reaction may also be seen in polymorphic light eruption. A mild lichenoid reaction with pigment incontinence was seen in one case of immunosseous dysplasia (OMIM 242900), which is caused by mutations in the SMARCAL1 gene. 1801
Magro and Crowson have reported 40 cases of lichenoid inflammation with a granulomatous component. 34 A drug was implicated in 14 cases. Over one-third of these patients with drug-related eruptions had other medical illnesses associated with cutaneous granulomatous inflammation, such as rheumatoid arthritis, Crohn’s disease, and hepatitis C. 34 A microbial trigger was implicated in 12 patients in the context of infective ‘id’ reactions to viral, fungal, or bacterial diseases. Hepatobiliary disease, rheumatoid arthritis, and cutaneous T-cell lymphoma were other associations. 34 The drugs included antibiotics, lipid-lowering agents, ACE inhibitors, and anti-inflammatory drugs. 34
A lichenoid and granulomatous reaction has since been reported in a patient presenting with erythroderma resulting from erythropoietin. 1802 The author has seen a similar pattern produced by allopurinol.
Breza and Magro have also reported three cases of this combined pattern in three patients with atypical (non-tuberculous) mycobacterial infection. 1803
References
Introduction
1. Pinkus, H, Lichenoid tissue reactions, Arch Dermatol 107 (1973) 840–846.
2. Weedon, D, The lichenoid tissue reaction, Int J Dermatol 21 (1982) 203–206.
3. Patterson, JW, The spectrum of lichenoid dermatitis, J Cutan Pathol 18 (1991) 67–74.
4. Weedon, D, Apoptosis in lichen planus, Clin Exp Dermatol 5 (1980) 425–430.
5. Weedon, D; Searle, J; Kerr, JFR, Apoptosis. Its nature and implications for dermatopathology, Am J Dermatopathol 1 (1979) 133–144.
6. Weedon, D, Apoptosis, Adv Dermatol 5 (1990) 243–256.
7. LeBoit, PE, Interface dermatitis. How specific are its histopathologic features? Arch Dermatol 129 (1993) 1324–1328.
8. Crowson, AN; Magro, CM; Mihm Jr, MC, Interface dermatitis, Arch Pathol Lab Med 132 (2008) 652–666.
9. Baima, B; Sticherling, M, How specific is the TUNEL reaction? An account of a histochemical study on human skin, Am J Dermatopathol 24 (2002) 130–134.
10. Hashimoto, K, Apoptosis in lichen planus and several other dermatoses, Acta Derm Venereol 56 (1976) 187–210.
11. Grubauer, G; Romani, N; Kofler, H; et al., Apoptotic keratin bodies as autoantigen causing the production of IgM-anti-keratin intermediate filament autoantibodies, J Invest Dermatol 87 (1986) 466–471.
12. Campisi, J, The role of cellular senescence in skin aging, J Invest Dermatol (Symposium Proceedings) 3 (1998) 1–5.
13. Haake, AR; Roublevskaia, I; Cooklis, M, Apoptosis: a role in skin aging? J Invest Dermatol (Symposium Proceedings) 3 (1998) 28–35.
14. Afford, S; Randhawa, S, Apoptosis, J Clin Pathol: Mol Pathol 53 (2000) 55–63.
15. Edwards, MJ; Jones, DW, Programmed cell death in human acute cutaneous wounds, J Cutan Pathol 28 (2001) 151–155.
16. Norris, DA, Differential control of cell death in the skin, Arch Dermatol 131 (1995) 945–948.
17. Pileri, S; Poggi, S; Sabattini, E; et al., Apoptosis as programmed cell death (PCD): Cupio dissolvi in cell life, Curr Diagn Pathol 1 (1994) 48–55.
18. Kerr, JFR; Winterford, CM; Harmon, BV, Apoptosis. Its significance in cancer and cancer therapy, Cancer 73 (1994) 2013–2026.
19. Raskin, CA, Apoptosis and cutaneous biology, J Am Acad Dermatol 36 (1997) 885–896.
20. Cummings, MC; Winterford, CM; Walker, NI, Apoptosis, Am J Surg Pathol 21 (1997) 88–101.
21. Chiodino, C; Cesinaro, AM; Ottani, D; et al., Expression of the novel inhibitor of apoptosis survivin in normal and neoplastic skin, J Invest Dermatol 113 (1999) 415–418.
22. Godar, DE, Light and death: photons and apoptosis, J Invest Dermatol (Symposium Proceedings) 4 (1999) 17–23.
23. Leonard, JV; Schapira, AHV, Mitochondrial respiratory chain disorders II: neurodegenerative disorders and nuclear gene defects, Lancet 355 (2000) 389–394.
24. Sellheyer, K; Krahl, D; Ratech, H, Distribution of Bcl-2 and Bax in embryonic and fetal human skin. Antiapoptotic and proapoptotic proteins are differentially expressed in developing skin, Am J Dermatopathol 23 (2001) 1–7.
25. Smith, KJ; Diwan, H; Skelton, H, Death receptors and their role in dermatology, with particular focus on tumor necrosis factor-related apoptosis-inducing ligand receptors, Int J Dermatol 42 (2003) 3–17.
26. Ständer, S; Schwarz, T, Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is expressed in normal skin and cutaneous inflammatory diseases, but not in chronically UV-exposed skin and non-melanoma skin cancer, Am J Dermatopathol 27 (2005) 116–121.
27. Birch-Machin, MA, The role of mitochondria in ageing and carcinogenesis, Clin Exp Dermatol 31 (2006) 548–552.
28. Malmusi, M; Ackerman, AB, Apoptosis is a type of necrosis. Part 1 – The importance of historical perspective and of definition, Dermatopathology: Practical & Conceptual 4 (1998) 15–27.
29. Bennion, SD; Middleton, MH; David-Bajar, KM; et al., In three types of interface dermatitis, different patterns of expression of intercellular adhesion molecule-1 (ICAM-1) indicate different triggers of disease, J Invest Dermatol 105 (1995) 71S–79S.
30. Oliver, GF; Winkelmann, RK; Muller, SA, Lichenoid dermatitis: A clinicopathologic and immunopathologic review of sixty-two cases, J Am Acad Dermatol 21 (1989) 284–292.
31. Godar, DE, UVA1 radiation triggers two different final apoptotic pathways, J Invest Dermatol 112 (1999) 3–12.
32. Okamoto, H; Mizuno, K; Itoh, T; et al., Evaluation of apoptotic cells induced by ultraviolet light B radiation in epidermal sheets stained by the TUNEL technique, J Invest Dermatol 113 (1999) 802–807.
33. Gilchrest, BA; Soter, NA; Stoff, JS; Mihm Jr, MC, The human sunburn reaction: histologic and biochemical studies, J Am Acad Dermatol 5 (1981) 411–422.
34. Magro, CM; Crowson, AN, Lichenoid and granulomatous dermatitis, Int J Dermatol 39 (2000) 126–133.
Lichenoid (interface) dermatoses
35. Boyd, AS; Neldner, KH, Lichen planus, J Am Acad Dermatol 25 (1991) 593–619.
36. Marshman, G, Lichen planus, Australas J Dermatol 39 (1998) 1–13.
37. Sánchez-Pérez, J; Rios Buceta, L; Fraga, J; García-Díez, A, Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients, Br J Dermatol 142 (2000) 310–314.
38. Karakatsanis, G; Patsatsi, A; Kastoridou, C; Sotiriadis, D, Palmoplantar lichen planus with umbilicated papules: an atypical case with rapid therapeutic response to cyclosporin, J Eur Acad Dermatol Venereol 21 (2007) 1006–1007.
39. Aytekin, S; Turhanoglu, AD; Ozkan, M; Uzunlar, AK, Clenched fist syndrome with palmar lichen planus, Int J Dermatol 44 (2005) 240–242.
40. Jobard-Drobacheff, C; Blanc, D; Quencez, E; et al., Lichen planus of the oesophagus, Clin Exp Dermatol 13 (1988) 38–41.
41. Evans, AV; Fletcher, CL; Owen, WJ; Hay, RJ, Oesophageal lichen planus, Clin Exp Dermatol 25 (2000) 36–37.
42. Itin, PH; Schiller, P; Gilli, L; Buechner, SA, Isolated lichen planus of the lip, Br J Dermatol 132 (1995) 1000–1002.
43. Lewis, FM; Shah, M; Harrington, CI, Vulval involvement in lichen planus: a study of 37 women, Br J Dermatol 135 (1996) 89–91.
44. Lewis, FM, Vulval lichen planus, Br J Dermatol 138 (1998) 569–575.
45. Vogel, PS; James, WD, Lichen planus of the eyelid: An unusual clinical presentation, J Am Acad Dermatol 27 (1992) 638–639.
46. Shurman, D; Reich, HL; James, WD, Lichen planus confined to a radiation field: The ‘isoradiotopic’ response, J Am Acad Dermatol 50 (2004) 482–483.
47. Kim, JH; Krivda, SJ, Lichen planus confined to a radiation therapy site, J Am Acad Dermatol 46 (2002) 604–605.
48. Shemer, A; Weiss, G; Trau, H, Wolf’s isotopic response: a case of zosteriform lichen planus on the site of healed herpes zoster, J Eur Acad Dermatol Venereol 15 (2001) 445–447.
49. Peluso, AM; Tosti, A; Piraccini, BM; Cameli, N, Lichen planus limited to the nails in childhood: case report and literature review, Pediatr Dermatol 10 (1993) 36–39.
50. Tosti, A; Peluso, AM; Fanti, PA; Piraccini, BM, Nail lichen planus: clinical and pathologic study of twenty-four patients, J Am Acad Dermatol 28 (1993) 724–730.
51. Tosti, A; Piraccini, BM; Cambiaghi, S; Jorizzo, M, Nail lichen planus in children. Clinical features, response to treatment, and long-term follow-up, Arch Dermatol 137 (2001) 1027–1032.
52. Richert, B; Iorizzo, M; Tosti, A; André, J, Nail bed lichen planus associated with onychopapilloma, Br J Dermatol 156 (2007) 1071–1072.
53. Colver, GB; Dawber, RPR, Is childhood idiopathic atrophy of the nails due to lichen planus? Br J Dermatol 116 (1987) 709–712.
54. Evans, AV; Roest, MAB; Fletcher, CL; et al., Isolated lichen planus of the toe nails treated with oral prednisolone, Clin Exp Dermatol 26 (2001) 412–414.
55. Lutz, M; Perniciaro, C; Lim, K, Zosteriform lichen planus without evidence of herpes simplex or varicella-zoster by polymerase chain reaction: report of two cases, J Cutan Pathol 24 (1997) 109.
56. Bansal, R, Segmental lichen planus, Int J Dermatol 43 (2004) 985.
57. Möhrenschlager, M; Engst, R; Hein, R; Ring, J, Primary manifestation of a zosteriform lichen planus: isotopic response following herpes zoster sine herpete? Br J Dermatol 158 (2008) 1145–1146.
58. Ott, H; Frank, J; Poblete-Gutiérrez, P, Eruptive lichen planus in a child, Pediatr Dermatol 24 (2007) 637–639.
59. Irvine, C; Irvine, F; Champion, RH, Long-term follow-up of lichen planus, Acta Derm Venereol 71 (1991) 242–244.
60. Mahood, JM, Familial lichen planus. A report of nine cases from four families with a brief review of the literature, Arch Dermatol 119 (1983) 292–294.
61. Kofoed, ML; Wantzin, GL, Familial lichen planus, J Am Acad Dermatol 13 (1985) 50–54.
62. Graells, J; Notario, J; Badia, F, Lichen planus in monozygotic twins, Clin Exp Dermatol 23 (1998) 299.
63. Sandhu, K; Handa, S; Kanwar, AJ, Familial lichen planus, Pediatr Dermatol 20 (2003) 186.
64. Powell, FC; Rogers, RS; Dickson, ER; Moore, SB, An association between HLA DR1 and lichen planus, Br J Dermatol 114 (1986) 473–478.
65. Carrozzo, M; Francia di Celle, P; Gandolfo, S; et al., Increased frequency of HLA-DR6 allele in Italian patients with hepatitis C virus-associated oral lichen planus, Br J Dermatol 144 (2001) 803–808.
66. Cottoni, F; Ena, P; Tedde, G; Montesu, MA, Lichen planus in children: a case report, Pediatr Dermatol 10 (1993) 132–135.
67. Milligan, A; Graham-Brown, RAC, Lichen planus in children – a review of six cases, Clin Exp Dermatol 15 (1990) 340–342.
68. Kamvar, AJ; Handa, S; Ghosh, S; Kaur, S, Lichen planus in childhood: a report of 17 patients, Pediatr Dermatol 8 (1991) 288–291.
69. Sharma, R; Maheshwari, V, Childhood lichen planus: a report of fifty cases, Pediatr Dermatol 16 (1999) 345–348.
70. Aste, N; Pau, M; Ferreli, C; Biggio, P, Lichen planus in a child requiring circumcision, Pediatr Dermatol 14 (1997) 129–130.
71. Nanda, A; Al-Ajmi, HS; Al-Sabah, H; et al., Childhood lichen planus: a report of 23 cases, Pediatr Dermatol 18 (2001) 1–4.
72. Kyriakis, KP; Terzoudi, S; Palamaras, I; et al., Sex and age distribution of patients with lichen planus, J Eur Acad Dermatol Venereol 20 (2006) 625–626.
73. García, RG; Castrillón, JLP; Ramón, SS; Romero, MP, Lichen planus in children and adolescents: a report of eight cases, J Eur Acad Dermatol Venereol 19 (2005) 265–267.
74. Handa, S; Sahoo, B, Childhood lichen planus: a study of 87 cases, Int J Dermatol 41 (2002) 423–427.
75. Luis-Montoya, P; Domínguez-Soto, L; Vega-Memije, E, Lichen planus in 24 children with review of the literature, Pediatr Dermatol 22 (2005) 295–298.
76. Balasubramaniam, P; Ogboli, M; Moss, C, Lichen planus in children: review of 26 cases, Clin Exp Dermatol 33 (2008) 457–459.
77. Flamenbaum, HS; Safai, B; Siegal, FP; Pahwa, S, Lichen planus in two immunodeficient hosts, J Am Acad Dermatol 6 (1982) 918–920.
78. Helm, TN; Camisa, C; Liu, AY; et al., Lichen planus associated with neoplasia: a cell-mediated immune response to tumor antigens? J Am Acad Dermatol 30 (1994) 219–224.
79. Gibson, GE; Murphy, GM, Lichen planus and carcinoid tumour, Clin Exp Dermatol 22 (1997) 180–182.
80. Calista, D, Oral erosive lichen planus associated with thymoma, Int J Dermatol 40 (2001) 762–764.
81. Epstein, O, Lichen planus and liver disease, Br J Dermatol 111 (1984) 473–475.
82. Rebora, A, Lichen planus and the liver, Int J Dermatol 31 (1992) 392–395.
83. Vainio, E; Huovinen, S; Liutu, M; et al., Peptic ulcer and Helicobacter pylori in patients with lichen planus, Acta Derm Venereol 80 (2000) 427–429.
84. Jubert, C; Pawlotsky, J-M; Pouget, F; et al., Lichen planus and hepatitis C virus-related chronic active hepatitis, Arch Dermatol 130 (1994) 73–76.
85. Cribier, B; Garnier, C; Laustriat, D; Heid, E, Lichen planus and hepatitis C virus infection: an epidemiologic study, J Am Acad Dermatol 31 (1994) 1070–1072.
86. Sanchez-Perez, J; De Castro, M; Buezo, GF; et al., Lichen planus and hepatitis C virus: prevalence and clinical presentation of patients with lichen planus and hepatitis C virus infection, Br J Dermatol 134 (1996) 715–719.
87. Chuang, T-Y; Stitle, L; Brashear, R; Lewis, C, Hepatitis C virus and lichen planus: a case-control study of 340 patients, J Am Acad Dermatol 41 (1999) 787–789.
88. Mignogna, MD; Muzio, LL; Favia, G; et al., Oral lichen planus and HCV infection: a clinical evaluation of 263 cases, Int J Dermatol 37 (1998) 575–578.
89. Bellman, B; Reddy, R; Falanga, V, Generalized lichen planus associated with hepatitis C virus immunoreactivity, J Am Acad Dermatol 35 (1996) 770–772.
90. Mignogna, MD; Muzio, LL; Russo, LL; et al., Oral lichen planus: different clinical features in HCV-positive and HCV-negative patients, Int J Dermatol 39 (2000) 134–139.
91. Tucker, SC; Coulson, IH, Lichen planus is not associated with hepatitis C virus infection in patients from North West England, Acta Derm Venereol 79 (1999) 378–379.
92. Jury, CS; Munro, CS, Linear lichen planus related to hepatitis C infection? Br J Dermatol 142 (2000) 836–837.
93. Daramola, OOM; George, AO; Ogunbiyi, AO, Hepatitis C virus and lichen planus in Nigerians: any relationship? Int J Dermatol 41 (2002) 217–219.
94. Amer, MA; El-Harras, M; Attwa, E; Raslan, S, Lichen planus and hepatitis C virus prevalence and clinical presentation in Egypt, J Eur Acad Dermatol Venereol 21 (2007) 1259–1260.
95. Aubin, F; Angonin, R; Humbert, P; Agache, P, Lichen planus following hepatitis B vaccination, Arch Dermatol 130 (1994) 1329–1330.
96. Saywell, CA; Wittal, RA; Kossard, S, Lichenoid reaction to hepatitis B vaccination, Australas J Dermatol 38 (1997) 152–154.
97. Ferrando, MF; Doutre, MS; Beylot-Barry, M; et al., Lichen planus following hepatitis B vaccination, Br J Dermatol 139 (1998) 350.
98. Schupp, P; Vente, C, Lichen planus following hepatitis B vaccination, Int J Dermatol 38 (1998) 799–800.
99. Rebora, A; Rongioletti, F; Drago, F; Parodi, A, Lichen planus as a side effect of HBV vaccination, Dermatology 198 (1999) 1–2.
100. Usman, A; Kimyai-Asadi, A; Stiller, MJ; Alam, M, Lichenoid eruption following hepatitis B vaccination: first North American case report, Pediatr Dermatol 18 (2001) 123–126.
101. Al-Khenaizan, S, Lichen planus occurring after hepatitis B vaccination: A new case, J Am Acad Dermatol 45 (2001) 614–615.
102. Limas, C; Limas, CJ, Lichen planus in children: a possible complication of hepatitis B vaccines, Pediatr Dermatol 19 (2002) 204–209.
103. Miteva, L, Bullous lichen planus with nail involvement induced by Hepatitis B vaccine in a child, Int J Dermatol 44 (2005) 142–144.
104. Calista, D; Morri, M, Lichen planus induced by hepatitis B vaccination: a new case and review of the literature, Int J Dermatol 43 (2004) 562–564.
105. De Vries, HJC; van Marle, J; Teunissen, MBM; et al., Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells, Br J Dermatol 154 (2006) 361–364.
106. Tasanen, K; Renko, M; Kandelberg, P; et al., Childhood lichen planus after simultaneous measles-mumps-rubella and diphtheria-tetanus-pertussis-polio vaccinations, Br J Dermatol 158 (2008) 646–648.
107. Manolache, L; Seceleanu-Petrescu, D; Benea, V, Lichen planus patients and stressful events, J Eur Acad Dermatol Venereol 22 (2008) 437–441.
108. Sardana, K; Sharma, RC; Koranne, RV; Mahajan, S, An interesting case of colocalization of segmental lichen planus and vitiligo in a 14-year-old boy, Int J Dermatol 41 (2002) 508–509.
109. Neumann-Jensen, B; Worsaae, N; Dabelsteen, E; Ullman, S, Pemphigus vulgaris and pemphigus foliaceus coexisting with oral lichen planus, Br J Dermatol 102 (1980) 585–590.
110. Creamer, D; McGregor, JM; McFadden, J; Hawk, JLM, Lichenoid tissue reaction in porphyria cutanea tarda, Br J Dermatol 141 (1999) 123–126.
111. Pretel, M; España, A, Lichen planus induced by radiotherapy, Clin Exp Dermatol 32 (2007) 582–583.
112. Gruppo Italiano Studi Epidemiologici in Dermatologia, Epidemiological evidence of the association between lichen planus and two immune-related diseases. Alopecia areata and ulcerative colitis, Arch Dermatol 127 (1991) 688–691.
113. Vassallo, C; Brazzelli, V; Martinoli, S; et al., Chronic Giardia intestinalis infection presenting with clinical features mimicking lichen planus, Acta Derm Venereol 81 (2001) 309–310.
114. Terheyden, P; Hornschuh, B; Karl, S; et al., Lichen planus associated with Becker’s nevus, J Am Acad Dermatol 38 (1998) 770–772.
115. Connelly, MG; Winkelmann, RK, Coexistence of lichen sclerosus, morphea, and lichen planus. Report of four cases and review of the literature, J Am Acad Dermatol 12 (1985) 844–851.
116. Rahnama, Z; Esfandiarpour, I; Farajzadeh, S, The relationship between lichen planus and hepatitis C in dermatology outpatients in Kerman, Iran, Int J Dermatol 44 (2005) 746–748.
117. Erkek, E; Bozdogan, Ö; Olut, AI, Hepatitis C virus infection prevalence in lichen planus: examination of lesional and normal skin of hepatitis C virus-infected patients with lichen planus for the presence of hepatitis C virus RNA, Clin Exp Dermatol 26 (2001) 540–544.
118. de Sousa Pinto, JM; Sacramento Marques, M; Estanislau Correia, T, Lichen planus and leukocytoclastic vasculitis induced by interferon alpha-2b in a subject with HCV-related chronic active hepatitis, J Eur Acad Dermatol Venereol 17 (2003) 193–195.
119. Harden, D; Skelton, H; Smith, KJ, Lichen planus associated with hepatitis C virus: No viral transcripts are found in the lichen planus, and effective therapy for hepatitis C virus does not clear lichen planus, J Am Acad Dermatol 49 (2003) 847–852.
120. Boyd, AS; Leonardi, CL, Absence of human papillomavirus infection in cutaneous lichen planus, J Am Acad Dermatol 36 (1997) 267–268.
121. Boyd, AS; Annarella, M; Rapini, RP; et al., False-positive polymerase chain reaction results for human papillomavirus in lichen planus. Potential laboratory pitfalls of this procedure, J Am Acad Dermatol 35 (1996) 42–46.
122. Yesudian, P; Rao, R, Malignant transformation of hypertrophic lichen planus, Int J Dermatol 24 (1985) 177–178.
123. Odukoya, O; Gallagher, G; Shklar, G, A histologic study of epithelial dysplasia in oral lichen planus, Arch Dermatol 121 (1985) 1132–1136.