35
Systemic Sclerosis and Sclerodermoid Disorders
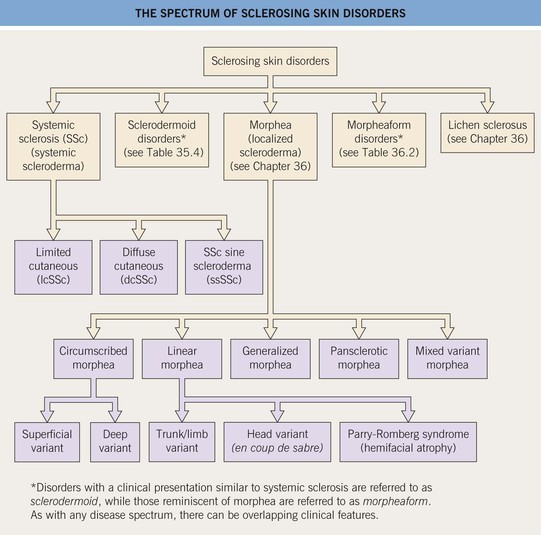
Systemic Sclerosis (SSc)
• Etiology unknown but pathogenesis involves vasculopathy, endothelial dysfunction, tissue fibrosis, and immune system activation.
• There are three major clinical subtypes of SSc, based on the amount of skin sclerosis (Fig. 35.2):
– Limited cutaneous SSc (lcSSc).
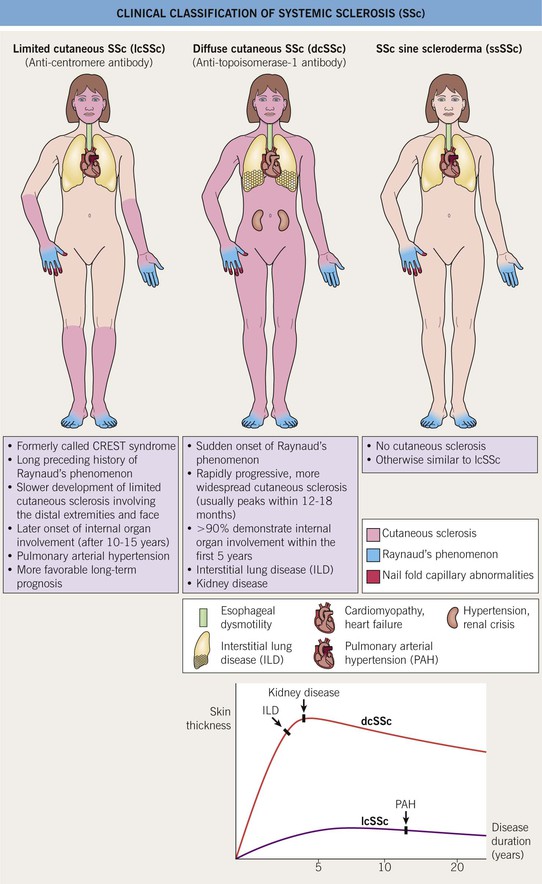
Fig. 35.2 Clinical classification of systemic sclerosis (SSc). In addition to the three major clinical subsets shown here, two others are recognized: pre-SSc, in which the full extent of the patient’s skin sclerosis has not been reached; and overlap syndrome, in which either lcSSc or dcSSc coexists with another AI-CTD, e.g. polymyositis or SLE. CREST, calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasias.
• Raynaud’s phenomenon (RP) is present in almost all SSc patients and is often the earliest presenting feature (Table 35.1; Figs. 35.3–35.5).
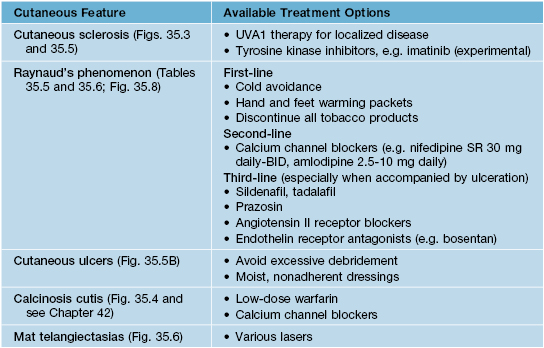
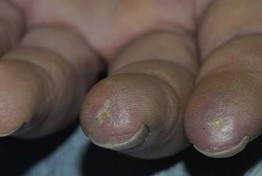
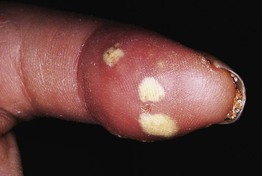
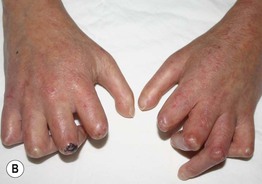
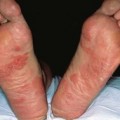
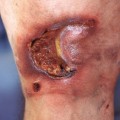
Fig. 35.5 Early versus late stages of systemic sclerosis (SSc) involving the hands. A Early edematous phase of SSc (note the demonstration of pitting edema on two of the digits). B Late stage of SSc with fixed flexion contractures, sclerodactyly, and digital ulceration overlying the third proximal interphalangeal joint. A, Courtesy, Jean L. Bolognia, MD; B, Courtesy, M. Kari Connolly, MD.

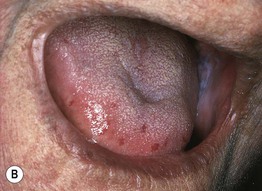
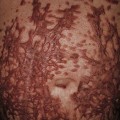
Fig. 35.6 Mat (squared-off) telangiectasias in two patients with systemic sclerosis. The first patient (A) had limited cutaneous systemic sclerosis (lcSSc, formerly called CREST syndrome) while the second patient (B) presented with diffuse hyperpigmentation and had interstitial lung disease. A, Courtesy, M. Kari Connolly, MD; B, Courtesy, Jean L. Bolognia, MD.
• Mat telangiectasias (Fig. 35.6) and proximal nailfold abnormalities (dilated capillary loops) are present in both lcSSc and dcSSc subtypes and are important clues to the Dx; additional common cutaneous findings are outlined in Table 35.1.
• Patients often have a characteristic facies with microstomia, retraction of the lips, perioral furrows, and a beaked nose; three types of dyspigmentation can also be seen, including diffuse hyperpigmentation and leukoderma of SSc (Fig. 35.7).
• In all three subtypes there can be internal organ involvement (Table 35.2), but patients with dcSSc are at increased risk for more clinically severe extracutaneous disease and overall worse outcomes.