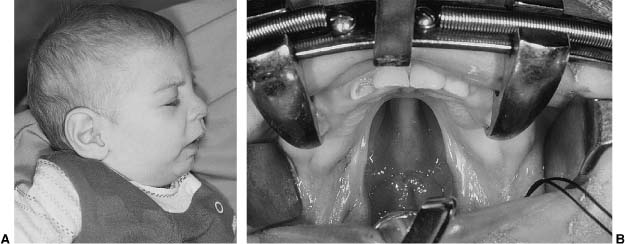
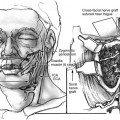
Chapter 1 Congenital anomalies are often complex, and may have significant physical and psychological impact. Successful management of the child with a congenital anomaly may require knowledge of genetics, pediatrics, molecular biology, and reconstructive surgery. An interdisciplinary team should include a speech therapist, social worker, pediatrician, geneticist, orthodontist, oral surgeon, audiologist, and cleft/craniofacial surgeon. Other specialists such as a neurosurgeon, ophthalmologist, or otologist may be necessary to provide the patient with comprehensive care. It is very important to address the psychosocial and genetic concerns of the patient and family. Although care of the patient with a cleft or craniofacial anomaly often demands a multidisciplinary effort, successful treatment is extremely rewarding. To understand human morphogenesis, a classification system for congenital anomalies is necessary. Most structural defects can be categorized into one of three general categories of developmental pathology: malformations, deformations, or disruptions (Table 1-1).1 The clinical manifestations and implications of each category of anomalies are different. A malformation is a morphologic defect of an organ, part of an organ, or a larger area of the body resulting from an intrinsically abnormal developmental process. A malformation sequence exists when there is a single localized poor formation of tissue that initiates a chain of subsequent defects. Malformation sequences occur in all types of expression, the manifestations ranging from nearly normal to extremely severe, and have a recurrence risk that is most commonly in the 1 to 5% range.2 Polydactyly, cleft lip, and renal agenesis serve as examples of malformation sequences. Malformations may be relatively simple or complex. The later in utero that a malformation defect is initiated, the simpler the malformation. Malformations initiated earlier in embryogenesis tend to have more significant consequences on the organism. A deformation is an abnormal form or position of a part of the body caused by nondisruptive mechanical forces. In a deformation sequence, there is no problem in the embryo or fetus, but mechanical forces such as uterine constraint result in altered morphogenesis, usually of the molding type. An example of a deformation sequence is oligohydramnios. This sequence is due to chronic leakage of amniotic fluid. Another deformation sequence is the breech deformation sequence, which occurs from prolonged breech position late in fetal life. Other deformations include clubfoot, congenital hip dislocation, and congenital postural scoliosis. Deformations usually arise during late fetal life and have a very good to excellent prognosis, in contrast with the prognosis of many malformation sequences. Because the most common cause of deformation is intrauterine molding by mechanical forces, the musculoskeletal system is usually affected. Deformations may result from mechanical, malformational, or functional causes. The recurrence risk for deformation is usually very low, unless the cause of the deformation problem is a persisting one, such as a bicornuate uterus.3 A disruption is a morphologic defect of an organ, part of an organ, or a larger region of the body resulting from a breakdown of, or interference with, an originally normal developmental process. In this scenario, a normal fetus is subjected to a destructive problem and its consequences. Disruptions may be of vascular, infectious, or even mechanical etiology. An example of a disruption sequence is the disruption of normally developing tissues by amnionic bands. An amputation of a digit in utero caused by an amnionic band or other band-caused disruptions such as ring constriction are other examples of disruption sequences. In uncommon cases, amniotic bands can cause dramatic manifestations such as asymmetric encephaloceles, bizarre facial clefting, and limb amputations. There is a fourth mechanism of abnormal morphogenesis. It is dysplasia, in which there is a lack of normal organization of cells into tissue.4 Hamartomas are examples of dysplasia sequence. This anomaly represents an abnormal organizational defect resulting in tumor-like formation of tissues. Some of these tissues have malignant potential. Examples of hamartomas are hemangiomas, melanomas, fibromas, lymphomas, adenomas, and other tumors that are not easily classified. Understanding of normal morphogenesis allows an interpretation of structural defects, and the study of structural defects aids in the understanding of normal morphogenesis. Each anomaly must have an understandable development and cause. Although distinctions among malformations, deformations, and disruptions are clinically useful in the newborn period, the three types of anomalies are somewhat interrelated during growth and development after birth. A sequence describes a pattern of structural defects in which all of the anomalies in the patient can be explained on the basis of a single problem in morphogenesis that leads to a cascade of subsequent defects. An example of such a sequence is the Robin sequence.5 The single initiating defect of this disorder may be hypoplasia of the mandibular region prior to 9 weeks in utero.6 The mandibular hypoplasia allows the tongue to be posteriorly located, thereby impairing the closure of the posterior palatal shelves that must overgrow the tongue to meet in the midline. This results in a U-shaped cleft palate. The resultant triad of mandibular hypoplasia, glossoptosis, and U-shaped cleft palate is referred to as the Robin sequence. This sequence may be malformational or deformational, depending on the initiating factor intrinsic mandibular hypoplasia (malformational) or extrinsic mandibular constraint (deformational). Although malformations, deformations, and disruptions can be distinguished as separate types of congenital anomalies, these classifications may be combined in some abnormalities. For example, some congenital malformations such as cleft lip and palate have secondary deformations. Although the original developmental anomaly is a malformation, the resulting secondary deformity occurs with growth and development of the organism. If a congenital abnormality is produced by the administration of a teratogen during pregnancy, the resulting facial abnormality is known as a deformation. This deformation occurs secondary to the original disruption created by the teratogen. The comparison of malformations, deformations, and disruptions usually does not have a significant affect on treatment philosophy. However, the distinction between these congenital anomaly types is important prognostically and in terms of relative recurrence risks. A sequence is defined as a single problem in morphogenesis that leads to a series of subsequence defects. Although often called syndromes, sequences are not true syndromes in the medical genetic sense. A syndrome is defined as multiple structural defects occurring in embryonically noncontiguous areas. These structural defects cannot be explained on the basis of a single initiating defect and its consequences, but rather appear to be the result of multiple defects in one or more tissues. An example of a syndrome in a child is congenital cleft lip and palate, genital malformations, and syndactyly of the toes. Although these malformations are embryonically noncontiguous, they are commonly thought to be due to a single cause. The known etiologies for malformation syndromes include chromosomal abnormalities, mutant gene disorders, and environmental teratogens. There are many syndromes for which the etiology has not been determined. Syndromes result from an interaction among genetic factors, molecular and cellular events, mechanical and deformational forces, and secondary effects of each of these on normal growth and development. A known syndrome is identified by a series of symptoms and signs that characterize an abnormal condition or disease state. Many malformation syndromes have distinctive facial appearances. Some of the dysmorphic features associated with these syndromes are easily identifiable, whereas others are difficult to describe or quantify. To become good at recognizing syndromes, physicians must train their eyes to recognize variations from normal, and must synthesize and collate these variations into recognizable patterns. The following sequences and syndromes include commonly recognizable craniofacial anomalies. The commonly seen triad of micrognathia, cleft palate, and glossoptosis was named after Pierre Robin,5 who reported the condition in 1923. However, the combination was described earlier by St. Hilaire in 1822, Fairbairn7 in 1846, and Shukowsky8 in 1911. The initiating defect of the Robin sequence (often mislabeled Pierre Robin syndrome) is mandibular hypoplasia occurring prior to 9 weeks’gestation. Latham9 studied a 17-week-old fetus with Robin sequence that had significant early mandibular retrognathia. The mandibular retrognathia associated with the Robin sequence pushes the tongue posteriorly and superiorly and impairs normal embryonic closure of the posterior palatal shelves, which normally close in an anterior-to-posterior fashion at between 8 and 12 weeks gestation.10 This closure begins anteriorly at the foramen cecum. The relative glossoptosis gives rise to a secondary U-shaped cleft palate. The extent of the palatal cleft is related to the degree of mandibular retrognathia (Fig. 1-1). Birth prevalence estimates have varied from 1/2000 to 1/30,000.11 Bush and Williams12 have suggested that the incidence of this sequence is 1/8500 births. Although the traditional definition of the Robin sequence includes the triad of micrognathia, cleft palate, and glossoptosis, Williams et al13 have cautioned against assuming that every infant with cleft palate and micrognathia has the sequence. In their review, they indicated that respiratory difficulty is an essential component of the Robin sequence. Although the Robin sequence is usually a malformation sequence, the condition can also result from early in utero mechanical constraint, with the chin being compressed in a manner to limit its growth prior to palatal closure. The manifestations of this deformation sequence are similar to the clinical condition of the malformation sequence. The Robin sequence can be associated with other congenital abnormalities and therefore be part of a syndrome (Table 1-2). Examples include Treacher Collins syndrome (mandibulofacial dysostosis, del [22q11.2] syndrome, and Stickler dysplasia syndrome).6 In syndromic forms of the Robin sequence, the mandible usually does not exhibit catch-up growth; rather, the mandible lags behind expected normal growth. In the deformational sequence form of the Robin sequence (due to intrauterine constraint), the mandible also does not catch up and grow to its expected size.14 These conditions are in contradistinction to the malformational form of Robin sequence, in which the mandible typically grows to normal size. Other syndromes in which Robin sequence may be a feature with variable frequency are listed in Table 1-3.15 In all cases of Robin sequence, the posterior airway obstruction must be carefully evaluated and treated. This may involve placing the child in the prone position, placing a nasopharyngeal airway, or doing more invasive procedures such as a lip-tongue adhesion or even a tracheotomy.16 The airway obstruction can be significant and lead to hypoxia, cor pulmonale, and occasionally failure to thrive. Affected children should be carefully monitored, especially during the first few weeks of life. FIGURE 1-1 (A) A 4-month-old girl with Pierre Robin sequence and obvious retrognathia. This patient also had relative macroglossia and a U-shaped cleft palate. (B) Close-up intraoperative view of a patient with U-shaped cleft palate. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.) The Treacher Collins syndrome was first reported in 1846 by Thompson17 and Toynbee18 and was later described by Treacher Collins19 in 1900. The more accurate term mandibulofacial dysostosis was first given to this syndrome by Franceschetti and Klein20 during the 1940s. This abnormality has several common features (Table 1-4). The constellation of abnormalities in Treacher Collins syndrome is easily recognizable by the significant mandibular hypoplasia and malar hypoplasia (Fig. 1-2). The zygomaticomalar hypoplasia results in a weakness in the lateral orbital rim and the associated soft tissues of the lower eyelid (Fig. 1-3). This causes an antimongoloid slant to the palpebral fissures of the eye. There are also commonly other first branchial arch defects, which include malformation of the auricles, defects of the external auditory canal, and conductive hearing loss. Occasional abnormalities include pharyngeal hypoplasia,21 coloboma of the upper eyelid, choanal atresia, and congenital heart defects. The occasional abnormalities associated with this syndrome are listed in Table 1-5.22
SYNDROMES AND CONGENITAL ANOMALIES
TYPES OF CONGENITAL ANOMALIES

RELATIONSHIPS BETWEEN TYPES OF ANOMALIES
SYNDROMES
ROBIN SEQUENCE
MANDIBULOFACIAL DYSOSTOSIS (TREACHER COLLINS SYNDROME, FRANCESCHETTI-ZWAHLEN-KLEIN SYNDROME)
| Condition |
|---|
| Monogenic |
| Beckwith-Wiedemann syndrome |
| Campomelic syndrome |
| Carey neuromuscular syndrome |
| Catel-Manzke syndrome |
| Cerebrocostomandibular syndrome |
| Congenital myotonic dystrophy |
| Diastrophic dysplasia |
| Donlan syndrome |
| Mandibulofacial dysostosis |
| Miller-Dieker syndrome |
| Nager acrofacial dysostosis |
| Otopalatodigital syndrome II |
| Persistent left superior vena cava syndrome |
| Popliteal pterygium syndrome |
| Postaxial acrofacial dysostosis |
| Radiohumeral synostosis syndrome |
| Robin-oligodactyly syndrome |
| Spondyloepiphyseal dysplasia congenita |
| Stickler syndrome |
| Velocardiofacial syndrome |
| Chromosomal |
| Del (4q) syndrome |
| Del (6q) syndrome |
| Dup (11q) syndrome |
| Teratogenically induced |
| Fetal alcohol syndrome |
| Fetal hydantoin syndrome |
| Fetal trimethadione syndrome |
| Disruption |
| Amniotic band sequence |
| Unknown genesis |
| CHARGE association |
| Femoral dysgenesis-unusual facies syndrome |
| Martsolf syndrome |
| Moebius sequence |
| Robin/amelia association |
| Sickle-shaped scapulae and club feet |
| Abnormal Chart | % |
|---|---|
| Antimongoloid slanting palpebral fissures | 89 |
| Malar hypoplasia, with or without cleft in zygomatic bone | 81 |
| Mandibular hypoplasia | 78 |
| Lower lid coloboma | 69 |
| Partial to total absence of lower eyelashes | 53 |
| Malformation of auricles | 77 |
| External ear canal defect | 36 |
| Conductive deafness | 40 |
| Visual loss | 37 |
| Cleft palate | 28 |
| Incompetent soft palate | 32 |
| Projection of scalp hair onto lateral cheek | 26 |
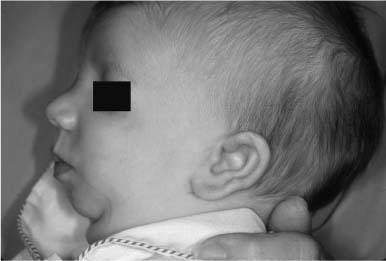
FIGURE 1-2 Lateral view of a 9-month-old with mandibular and malar hypoplasia and malformation of the auricles.
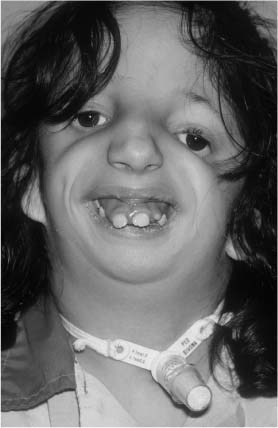
FIGURE 1-3 Patient with significant zygomaticomalar hypoplasia resulting in weakness of the lateral orbital rim and a secondary antimongoloid slant to the palpebral fissures of the eye.
The genetic etiology of Treacher Collins syndrome is autosomal dominant with 60% of the cases representing fresh mutations. The genetic map location for this abnormality is 5q32-33.1. Patients with Treacher Collins syndrome often develop respiratory problems as a result of the mandibular hypoplasia and narrow oropharyngeal airway. Occasionally, patients with mandibulofacial dysostosis require tracheotomy (Fig. 1-4). These patients should be carefully monitored and tested for hearing loss and for visual loss. Otologic and ophthalmologic consultation is recommended.
| Occasional Abnormal Chart |
|---|
| Pharyngeal hypoplasia |
| Coloboma of the upper lid |
| Dacrostenosis |
| Microphthalmia |
| Strabismus |
| Ptosis |
| Macrostomia |
| Microstomia |
| Choanal atresia |
| Blind fistulas and skin tags between auricle and angle of the mouth |
| Absence of the parotid gland |
| Congenital heart defect |
| Cryptorchidism |
| Mental deficiency 5% |
STICKLER SYNDROME (HEREDITARY ARTHRO-OPHTHALMOPATHY)(MARSHALL-STICKLER SYNDROME, WAGNER-STICKLER SYNDROME)
In 1965, Stickler23 observed five generations of one family with affected individuals, noting facial characteristics of flat facies and depressed nasal bridge with midfacial or mandibular hypoplasia. The complete manifestations of the Stickler syndrome were elaborated by Herrmann et al,24 who additionally noted myopia and spondyloepiphyseal dysplasia. Stickler syndrome consists of abnormalities in the orofacial region, the eyes, and the musculoskeletal system (Fig. 1-5). The facial abnormalities consist of flat facies, secondary cleft of the hard or soft palate (20%), Robin sequence, sensorineural and conductive hearing loss (80%), epicanthal folds, and midfacial or mandibular hypoplasia.25 These patients have significant myopia of 8 to 18 diopters (75-80%). This myopia is usually present before the age of 10 years. The patients also can have chorioretinal degeneration, retinal detachment, or cataracts.
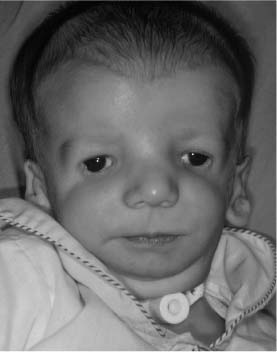
FIGURE 1-4 Patient with mandibulofacial dysostosis requiring tracheotomy. Note the coloboma of the lower eyelids, and the downward slanting of the poorly supported lateral canthi of the eyelids.
The musculoskeletal system in patients with Stickler syndrome can have various manifestations.26–28 These include hypotonia, hyperextensible joints, marfanoid body habitus, scoliosis, and other severe arthropathies. The patients can have subluxation of the hips and other joints in mild to moderate spondyloepiphyseal dysplasia. Secondary degeneration of the articular surface of joints can occur in adulthood. Other manifestations of Stickler syndrome include mitral valve prolapse, which occurs in 46% of patients.29
All patients found to have the Robin sequence at birth should be considered possible candidates for Stickler syndrome.30,31 It has been suggested that 30% of infants with Robin sequence are eventually diagnosed with Stickler syndrome. This is particularly true in any patient with a family history of cleft palate or in any patient with congenital myopia or retinal problems. Ophthalmologic consultation should be obtained at approximately 3 to 6 months of age, and the patient should get retested at about 1 year of age. The genetic etiology is autosomal-dominant inheritance with highly variable expression. The syndrome is noted to be secondary to a mutation of the type 2 collagen gene, COL2AI, which is located on a chromosome 12q13.11-q13.3.32

FIGURE 1-5 A 5-year-old girl with Stickler syndrome and significant myopia.
VELOCARDIOFACIAL SYNDROME (SHPRINTZEN SYNDROME, SEDLACKOVA SYNDROME, DELETION 22Q11.2)
In 1978, Shprintzen et al33 described a condition with prominent nose, retruded mandible, cardiovascular anomalies, cleft palate, and learning disabilities. As early as 1955, Sedlackova34 and coworkers reported similar patients and called the condition velofacial hypoplasia. The velocardiofacial syndrome has various manifestations,35 including cardiac (85%), palatal, auricular (60%), otologic (conductive hearing loss in 75%), and general intellect and developmental problems. Hypotonia and upper respiratory illnesses in infancy and childhood are frequent. Failure to thrive in infancy is present in ~25%, and ~35% manifest short stature. Learning disabilities are experienced by 100%.
Cardiac defects are present in 85% of patients, with the most common being the ventricular septal defect (65%).36 Other cardiac problems include a right-sided aortic arch (35%), tetralogy of Fallot (20%), and an aberrant left subclavian artery (20%). Craniofacial manifestations include clefts of the secondary palate, which involve either an overt cleft of the palate, a submucous cleft, or a dysfunction of the palatal muscles.37 This usually results in velopharyngeal insufficiency. Other common features include a prominent nose with a narrow alar base and flat nasal root. Most patients have vertical maxillary excess (85%) with a long face syndrome and mild retrusion of the mandible with microgenia (Fig. 1-6). Patients also have conductive hearing loss (75%) and auricular deformities, learning disabilities and low IQ, and hyper-extensible joints in the hands and fingers. Occasional abnormalities include abnormalities of the internal carotid artery, other types of cardiac defects not listed above, hypospadias, scoliosis, and abnormalities of the retinal vessels.38
The genetic inheritance of this syndrome is autosomal dominant. Individuals affected with Shprintzen syndrome have an interstitial deletion of chromosome 22q11.21-q11.23.39 Cytogenetic analysis can be performed with a fluorescent in situ hybridization (FISH) test. However, this analysis detects only 20% of the deletions in the region. It is important to note that chromosome 22q11 is in the same region that is deleted in cases of DiGeorge sequence.40 In that many patients have parents or siblings with both DiGeorge sequence and velocardiofacial syndrome, it is believed that the two congenital disorders represent different manifestations of the same genetic defect.
VAN DER WOUDE SYNDROME (LIP PIT-CLEFT LIP/PALATE SYNDROME)
The first report of congenital sinuses of the lower lip (Fig. 1-7) in combination with cleft lip and/or cleft palate appears to be by Demarquay in 1845.41 The syndrome was then independently reported by Murray42 in 1860 and got its name from the report by Van der Woude43 in 1954. The syndrome occurs in roughly 1 to 2% of patients with facial clefts or ~1:35,000 to 1:100,000 in the Caucasian population. The common abnormalities include cleft lip with or without cleft palate, cleft palate alone, cleft uvula, and lower lip pits, which are present in 80% of patients (Fig. 1-8). Patients can also have hypodontia and missing central and lateral incisors. The lower lip pits represent small accessory salivary glands.44–46
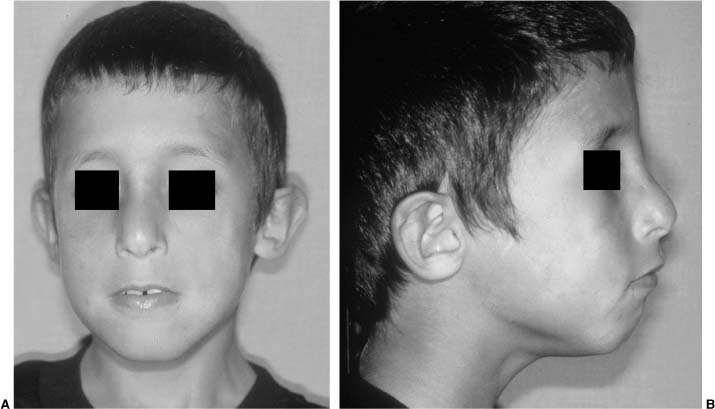
FIGURE 1-6 Frontal (A) and lateral (B) photographs of a child with velocardiofacial syndrome exhibiting mild mandibular retrusion, microgenia, and mild vertical maxillary excess. This patient also has a broad and flat nasal root.
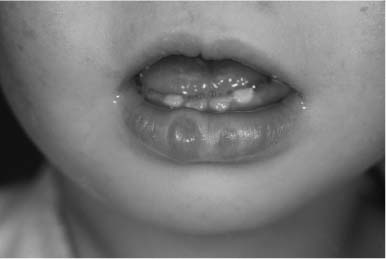
FIGURE 1-7 Child with Van der Woude syndrome and congenital sinuses of the lower lip.
The inheritance of the etiology syndrome is autosomal dominant. The responsible gene for Van der Woude syndrome has been mapped to the long arm of chromosome 1 at q32-41.47 Another deletion of 2q34-q36 may also be responsible for this syndrome.
OROFACIODIGITAL SYNDROME
Since the first description by Mohr48 in 1941, many cases of various types of orofaciodigital (OFD) syndrome have been described. Nine different specific syndromes have been delineated. The syndromes consist of oral frenula and oral clefts, hypoplasia of the nasal ala, conductive hearing loss, and multiple digital problems. The major abnormalities of OFD syndrome type 1 are listed in Table 1-6. OFD syndrome type 1 was first described by Papillon-Leage and Psaume49 in 1954. Approximately 50% of patients have mental deficiency. Many patients do poorly in early infancy with as many as one third of these dying in early childhood. The etiology of OFD syndrome type 1 is X-linked dominant with a vast majority of affected males dying. OFD syndrome type 2 (Mohr syndrome) consists of cleft tongue (Fig. 1-9), conductive hearing loss, and partial reduplication of hallux. The etiology of this syndrome (type 2) is autosomal recessive. These patients have normal intelligence.
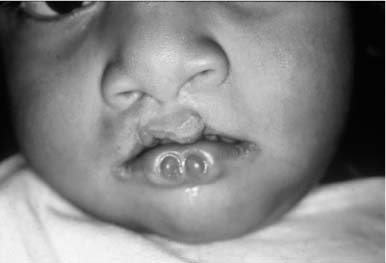
FIGURE 1-8 South American patient with previously repaired bilateral cleft lip, Van der Woude syndrome, and obvious large congenital lower lip pits.
| Common Abnormalities: Oral, Facial, Digital, Scalp, Central Nervous System, Cranium, Renal |
| Oral: Multiple and/or hyperplastic frenula between the buccal mucous membrane and alveolar ridge. Median cleft lip. |
| Lobated/bifid tongue with nodules. Cleft of alveolar ridge (at area of lateral incisors, which may be missing). Cleft palate. |
| Dental caries and anomalous anterior teeth. |
| Facial: Hypoplasia of alar cartilages, lateral placement of inner canthi. Milia of ears and upper face in infancy. |
| Digital: Asymmetric shortening of digits with clinodactyly, syndactyly, and/or brachydactyly of hands and unilateral polydactyly of feet. |
| Scalp: Dry, rough, sparse hair and dry scalp. |
| Central nervous system: Variable mental deficiency in ~57%, with average IQ of 70. Brain malformation, including absence of corpus callosum and heterotopia of gray matter, in ~20% of patients. |
| Cranium: Increased naso-sella-basion angle at base of cranium. |
| Renal: Adult polycystic kidney disease. |
| Occasional Abnormalities: Oral, Facial, Skeletal, Central Nervous System, Hair, Skin |
| Oral: Enamel hypoplasia, supernumerary teeth, hamartoma of tongue, fistula in lower lip. Choanal atresia. |
| Facial: Frontal bossing. Hypoplastic mandibular ramus and zygoma. |
| Skeletal: Nonprogressive metaphyseal rarefaction. |
| Central nervous system: Trembling, hydrocephalus, seizures, berry aneurysm. |
| Hair: Alopecia. |
| Skin: Granular seborrheic skin. |
OCULO-AURICULO-VERTEBRAL SPECTRUM (FIRST AND SECOND BRANCHIAL ARCH SYNDROME, FACIO-AURICULO-VERTEBRAL SPECTRUM, HEMIFACIAL MICROSOMIA, GOLDENHAR SYNDROME)
The oculo-auriculo-vertebral (OAV) spectrum, often called Goldenhar syndrome,50 represents abnormalities in morphogenesis of the first and second branchial arches.51 These defects are sometimes accompanied by vertebral or ocular anomalies. When accompanied by epibulbar dermoids (Fig. 1-10), this spectrum is designated as the Goldenhar syndrome. When the abnormality is primarily unilateral, the OAV spectrum is termed hemifacial microsomia (Fig. 1-11).52 The frequency of occurrence of the OAV spectrum is estimated to be 1 in 3000 to 1 in 5000 with a slight (3:2) male predominance.
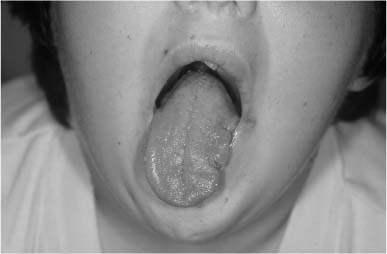
FIGURE 1-9 Close-up view of a patient with orofacial digital syndrome and lateral clefting of the tongue.
The OAV spectrum affects auricular, oral, and mandibular growth.53 Although bilateral involvement is common, frequently there is more severe expression of the disorder on one side, giving rise to significant facial asymmetry. The common abnormalities of the OAV spectrum are noted in Table 1-7. The occasional abnormalities of the spectrum are noted in Table 1-8.54

FIGURE 1-10 A 5-month-old with oculo-auriculo-vertebral (OAV) spectrum exhibiting colobomas of the upper and lower eye lids, epibulbar dermoids of the left lateral canphus, macrostomia, malformed auricles, and skin tags of the face. The patient also has facial asymmetry and hemifacial microsomia.

FIGURE 1-11 An 11-year-old girl with obvious facial asymmetry, hemifacial microsomia, and a clear occlusal cant.
Most cases of OAV spectrum are sporadic in etiology. However, familial instances can be observed, suggesting an autosomal-dominant inheritance pattern in some families. Within a given family, manifestations of the spectrum vary from severe involvement to simple auricular deformities or preauricular skin tags (Fig. 1-12). There is a 3:2 predilection for right-sided ear involvement.
| Facial: Hypoplasia of malar, maxillary, and/or mandibular region, especially ramus and condyle of mandible and temporomandibular joint. Lateral cleft-like extension of corner of mount (macrostomia). Hypoplasia of facial musculature. |
| Hypoplasia of depressor anguli oris. |
| Ear: Microtia, accessory preauricular tags and/or pits, most commonly in a line from the tragus to the corner of the mouth. Middle ear anomaly with variable deafness. |
| Oral: Diminished to absent parotid secretion. Anomalies in function or structure of tongue. Malfunction of soft palate. |
| Vertebral: Hemivertebrae or hypoplasia of vertebrae, most commonly cervical but may also be thoracic or lumbar. |
| Eye: Epibulbar dermoid, lipodermoid, notch in upper lid, strabismus, microphthalmia |
| Ear: Inner ear defect with deafness |
| Oral: Cleft lip, cleft palate |
| Cardiac: Ventricular septal defect, patent ductus arteriosus, tetralogy of Fallot, and coarctation of aorta, in decreasing order |
| Genitourinary: Ectopic and/or fused kidneys, renal agenesis, vesicoureteral reflux, ureteropelvic junction obstruction, ureteral duplication, and multicystic dysplastic kidney |
| Other: Mental deficiency (IQ below 85 in 13%), branchial cleft remnants in anterior-lateral neck, laryngeal anomaly, hypoplasia to aplasia of lung, hydrocephalus, Arnold-Chiari malformation, occipital encephalocele, agenesis of corpus callosum, calcification of falx cerebri, hypoplasia of septum pellucidum, intracranial dermoid cyst, lipoma in corpus callosum, radial and/or rib anomalies, prenatal growth deficiency, low scalp hairline |
The variability of OAV spectrum is significant. Some patients have an extensive range of anomalies, whereas others have only a single minor problem, such as a dysplastic ear (Fig. 1-13) or a small amount of facial asymmetry. Extensive family histories should be obtained for auricular and ocular anomalies, vertebral and skeletal problems, and cleft lip or cleft palate. Approximately 7% of reported patients have a cleft lip or cleft palate. Other features include mental deficiency (10%), vertebral abnormalities (40–60%), cardiovascular anomalies (45–55%), and a variety of other central nervous system anomalies. Most patients are of normal intelligence, although mental deficiency can occur. Many patients undergo cosmetic or reconstructive surgery to correct one or more of the auricular, ocular, or facial skeletal problems.
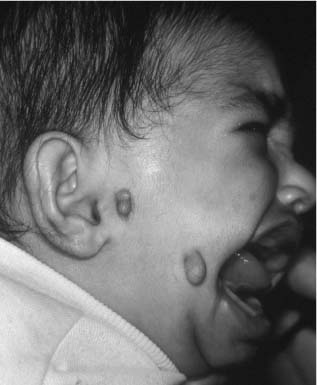
FIGURE 1-12 Patient with OAV spectrum, significant clefting of the lateral oral commissure, and multiple preauricular and facial skin tags.
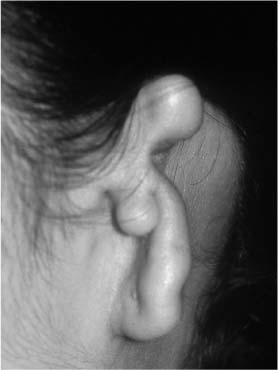
FIGURE 1-13 Patient with OAV spectrum and significant dysplasia of the ear.
OROFACIAL CLEFTING
Facial clefting represents the most common developmental abnormality of the face. There is great variability in the degree of cleft formation, with minimal involvement manifesting as a bifid uvula, microform clefts of the lip with minor indentations, lateral upper lip fistula, and submucous palatal clefts. Severe manifestations of facial clefting can also occur and include bilateral complete clefts of the lip and palate or lateral facial clefts, which involve the maxilla, zygoma, and orbit. The degree of clefting of the lip, palate, nose, and remainder of the face is related to the amount of embryologic insult. To better understand facial clefts and their related syndromes, the embryologic, epidemiologic, and genetic aspects of isolated cleft lip and cleft palate are reviewed in the following subsections.
EMBRYOLOGY OF THE LIP AND PALATE
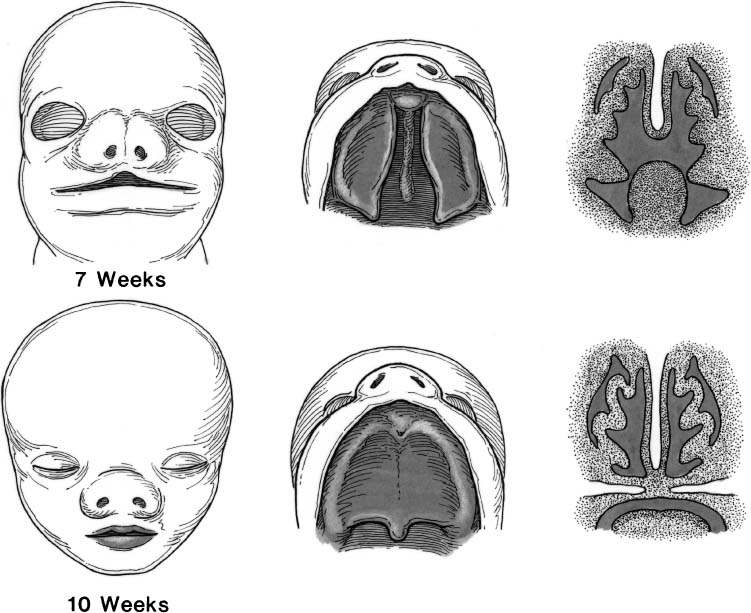
The critical period in the formation of the face is the embryonic period. During the first 4 to 12 weeks of embryogenesis, most developmental abnormalities occur. Normal development of the lip and palate occurs during the first 12 weeks of intrauterine life. The midportion of the face develops anterior to the forebrain by the differentiation of the broad midline frontonasal prominence (Fig. 1-14). The primary palate forms at approximately 4 to 6 weeks, and forms the initial separation between the oral and nasal cavities. The primary palate, or median palatine process, is formed by the fusion of the paired median nasal prominence (MNP). Fusion of the paired MNP gives rise to the central upper lip, the central maxillary alveolar arch and associated lateral and central incisors, and the hard palate anterior to the incisive foramen.55,56 Development of the primary palate (anterior to the incisive foramen) is distinct embryologically from normal formation of the secondary palate (posterior to the incisive foramen).
The paired median nasal processes coalesce during the sixth week and eventually form the premaxilla, philtrum of the upper lip, columella, and nasal tip (Fig. 1-15). The lateral elements of the upper lip (from the philtral column laterally) are derived from the paired maxillary processes. The cheek, maxilla, zygoma, and secondary palate are also formed from the maxillary processes. The upper lip, therefore, is formed from both the median nasal and maxillary processes.57
The secondary palate begins developing at approximately 8 weeks of gestation, after development of the primary palate is completed. Formation of the secondary palate occurs by inferior and medial growth and migration of the palatal shelves (the medial projections of the maxillary processes). As the palatal shelves displace inferiorly (similar to a drawbridge), the developing nasal cavities expand laterally and inferiorly.
The paired palatal shelves are initially separated by the developing tongue. Growth of the mandible with associated anterior movement of the tongue allows the palatal shelves to migrate inferiorly and assume a more horizontal orientation. If fetal development and migration of the mandible does not proceed normally, the paired palatal shelves cannot migrate inferiorly and medially. Lack of contact of these shelves creates palatal clefting. This malformation can result in the Pierre Robin sequence (micrognathia, relative macroglossia, and U-shaped cleft palate).58 The normal sequence of palatal formation begins when the palatal shelves and nasal septum contact each other, and proceeds in an anterior-to-posterior direction beginning at the incisive foramen. Palatal closure first occurs at the incisive foramen at approximately 8 weeks of gestation and is usually completed through the uvula by 12 weeks. The degree of clefting of the secondary palate is related to many factors including when in fetal development the fusion process is interrupted. Therefore, the palatal abnormality associated with the Pierre Robin sequence can be only a bifid uvula, a submucous soft palate cleft, or a complete cleft of the secondary palate.59
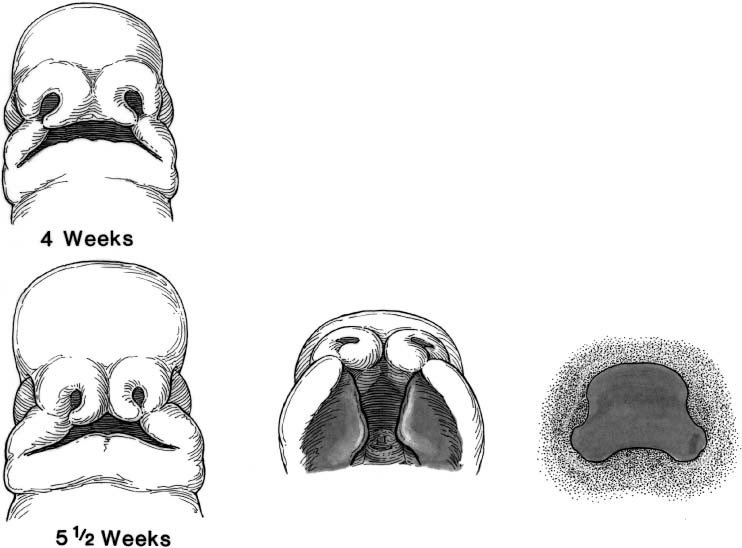
FIGURE 1-14 Schematic diagram of the development of the lip and palate at 4 and 5 1/2 weeks of gestational age. The primary palate is forming at about 4 to 6 weeks by fusion of the paired median nasal prominences. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.)
EPIDEMIOLOGY OF CLEFT LIP AND PALATE
Cleft Lip with or Without Cleft Palate
For the purposes of the epidemiology and genetics of facial clefting, congenital clefting can be divided into (1) cleft lip with or without cleft palate and (2) isolated cleft palate. The incidence of cleft lip with or without cleft palate is ~1/1000 white births.60,61 The relative incidence varies significantly according to race, with American Indians having the highest incidence (3.6/1000)62,63 compared with African-American births (0.3/1000).64,65 The incidence is also high in Japanese66 (~2.1/1000) and Chinese (1.7/1000) births.67,68
Isolated cleft lip without cleft palate is typically unilateral (80%),69 more commonly located on the left side (70%),70 and usually present in boys (60%). However, not all races have similar distributions of sex and sidedness. Ten percent of cleft lips are incomplete. Approximately 85% of cases of bilateral cleft lip and 70% of unilateral cleft lip are associated with cleft palate.
Isolated Cleft Palate
Isolated cleft palate appears to be a separate entity from cleft lip with or without cleft palate. The siblings of patients with cleft lip with or without cleft palate have an increased frequency of this anomaly, but these siblings do not have an increased incidence of isolated cleft palate.71 The incidence of isolated cleft palate in white births is 0.5/1000.72 Isolated palatal clefting is more common in girls (2:1), but the ratio becomes closer to 1:1 when clefts of only the soft palate are considered.73 Submucous clefts of the soft palate are caused by incomplete union of the fusing palatal muscles.74 The incidence of submucosal soft palate clefts is 1/1200 to 1/2000 white births with greater than 50% of these patients being boys.75,76 The overall incidence of cleft uvula (1/80 white births) is expectedly much higher than the incidence of cleft palate (1/2500 births).77 The sex preponderance for clefting of the uvula is 1:1.78 As opposed to complete and submucosal clefts, clefts of the uvula tend to vary with race, with American Indians having the highest incidence at 1:9 to 1:14 births.79,80
Lateral and oblique clefts as classified by Tessier and occur uncommonly. It is estimated that lateral facial clefts represent 0.25% of cleft births or 1 in 50,000 to 1 in 175,000 live births.81,82
Genetics of Facial Clefting
The etiology of facial clefting is multifactorial. Currently, there is no true consensus as to the genetic cause of facial clefting. The cause is likely a heterogeneous process that results from a variety of factors including major genes (autosomal dominant with incomplete penetrance, codominant, and autosomal recessive), minor genes, environmental insults, and developmental threshold. It is clear that the recurrence risks for facial clefting do not correspond to any simple mendelian pattern of inheritance. This fact is supported by various studies on twins who have facial clefts. Twin studies have identified the relative roles of both genetic and nongenetic factors on cleft production. Gorlin et al15,83 have tabulated twin studies in 1988 (Table 1-9) in twins with cleft lip with or without cleft palate; concordance is far greater for monozygotic (36%) than for dizygotic twins (4.7%). The fact that the concordance in monozygotic twins (36%) is not 100% suggests that there are factors other than genetics involved in clefting. In twins with isolated cleft palate, the difference in concordance between monozygotic (22%) twins and dizygotic (4.6%) twins is not as great. This suggests a stronger genetic basis for cleft lip with or without cleft palate than the genetic basis for isolated cleft palate. The concordance of 4.6% for dizygotic twins is greater than that for nontwin siblings, which implies that maternal factors are present in causing clefting.
If genetics were the only basis for clefting, monozygotic twins would have a 100% concordance, whereas dizygotic twins who share up to 50% of their genes would have a higher concordance than is demonstrated in these twin studies. The largest twin study demonstrated concordance for monozygotic twins for cleft palate of only 50% and in dizygotic twins of 0%. Conclusions from all twin studies indicate that both genetic and environmental factors influence the development of facial clefting, and there seems to be a stronger genetic influence on cleft lip with or without cleft palate than there is on cleft palate alone.
Environmental factors have been proven to have a direct effect on the incidence of clefting. Phenytoin, retinoic acid derivatives, and folic acid antagonists have all been shown to have a direct relation with clefting. Cigarette smoking with its relative fetal hypoxia and first-trimester alcohol use have also been implicated as factors that increase the risk of clefting. Recent data indicate that folic acid supplements taken by mothers during the embryonic stage of development can reduce the incidence of nonsyndromic facial clefting.
The final genetic point is that facial clefting occurs within syndromes and outside of syndromes. A syndrome must include a cognitive or structural abnormality that is distinct from the cleft; the abnormalities are thought to be pathologically related; 30 to 65% of facial clefts are related to a named syndrome. Cleft lip with or without cleft palate is associated with syndromes 14% of the time, and cleft palate alone is associated with a syndrome nearly 55% of the time. There are more than 300 named syndromes that include facial clefting.

Recurrence Risks for Facial Clefting
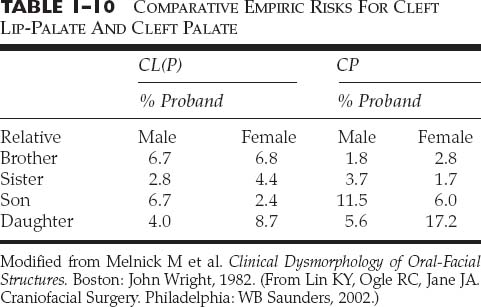
The relative recurrence risks for cleft lip with or without cleft palate and for isolated cleft palate for an affected parent or in the case of normal parents who have an affected child have been determined in numerous studies. For cleft lip with or without cleft palate and for isolated cleft palate, if the proband has no other affected first- or second-degree relatives, the empiric risk is 3 to 5%. However, if the affected child has other affected first-degree relatives, the risk to unborn siblings rises to 10 to 20%.84,85 In the uncommon case of both parents being affected with facial clefting, the empiric risk data range between 25 and 50% (Table 1-10). For her Ph.D. thesis, Tolarová calculated the relative recurrence risks for unborn offspring with mother, father and affected sibs (Table 1-11).
Classification of Clefts
The embryologic development of the lip and palate serves as a natural mechanism to classify congenital clefts. The upper lip and primary palate develop at between 4 and 8 weeks of gestation. Interruption of this development can cause clefts of the upper lip and alveolus (primary palate). Cleft lips may be unilateral or bilateral, and may be complete or incomplete. The upper lip clefting may be isolated, may be associated with clefts of the alveolus and palate, or may be associated with other malformations (syndromic clefts). Clefting of the upper lip, with or without associated palatal clefting, is caused by failure of the median nasal prominences to make contact with the lateral nasal process and the maxillary process during embryogenesis. Interruption of this embryologic process creates maldevelopment of some or all of the upper lip, the central maxillary arch, the anterior portion of the palate, and the base of the nose and nasal tip.
| Affected Parent | CL(P) % | CP % |
|---|---|---|
| Mother affected | ||
| Affected sibs | ||
| 0 | 2.7 | 2.3 |
| 1 | 9.9 | 11.2 |
| 2 | 18.3 | 21.1 |
| Father affected | ||
| Affected sibs | ||
| 0 | 2.3 | 5.0 |
| 1 | 9.3 | 14.4 |
| 2 | 17.6 | 23.9 |
| Both parents affected | ||
| Affected sibs | ||
| 0 | 24.0 | 45.0 |
| 1 | 31.7 | 51.6 |
| 2 | 37.6 | 54.5 |
From Tolarová, Ph.D. Thesis, Charles University, Prague, Czechoslovakia, 1984. (From Lin KY, Ogle RC, Jane JA. Craniofacial Surgery. Philadelphia: WB Saunders, 2002.)
The degree of clefting of the upper lip, alveolus, and nose is related to the amount of the embryologic insult to the upper lip. The lip deformity ranges from a minor malformation of the normal development of the lip to a complete interruption of all layers of the lip and base of the nose. A minor malformation of the normal lip development may cause dehiscence of the orbicularis oris muscle with no overt clefting of the epidermis of the upper lip. This condition is known as a microform cleft of the upper lip (Fig. 1-16). A more significant interruption of the upper lip development is referred to as an incomplete cleft lip (Fig. 1-17). An incomplete unilateral cleft lip involves a through-and-through defect of skin, muscle, and mucosa of the lower aspect of the lip, but does not extend superiorly through the entire height of the lip. A complete unilateral clef lip occurs when the defect involves all tissue layers of the upper lip and extends through the entire height of the lip and floor of the nose (Fig. 1-18). A cleft of the alveolus is almost always associated with complete clefting of the upper lip.
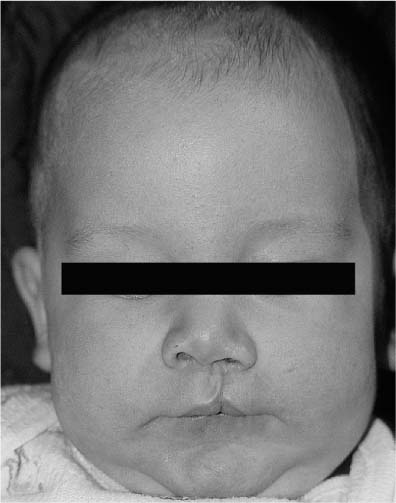
FIGURE 1-16 A 3-month-old with a left microform cleft lip. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.)
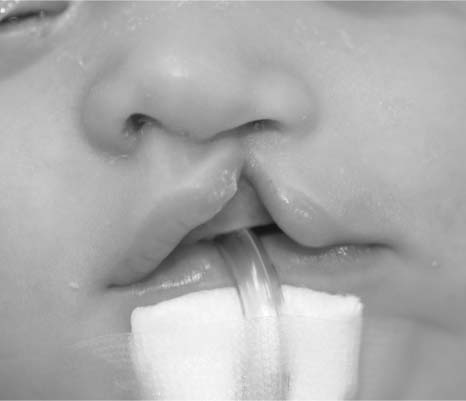
FIGURE 1-17 A 3-month-old with an incomplete left cleft lip. Note the alar base asymmetry and inferior displacement of the left alar rim.
The degree of clefting of the upper lip depends on the timing and degree of interruption of normal lip development. If lip development of both sides is disrupted, a bilateral cleft lip results. In the incomplete bilateral cleft lip, there is usually some skeletal continuity between the lateral maxillary processes and the central premaxilla (Fig. 1-19). For this reason, there is often little or no protrusion of the premaxilla in incomplete bilateral cleft lips. In the complete bilateral cleft lip deformity, the central premaxilla is totally detached from each lateral maxillary process (Fig. 1-20). This may result in a “locked out” premaxilla (Fig. 1-21).
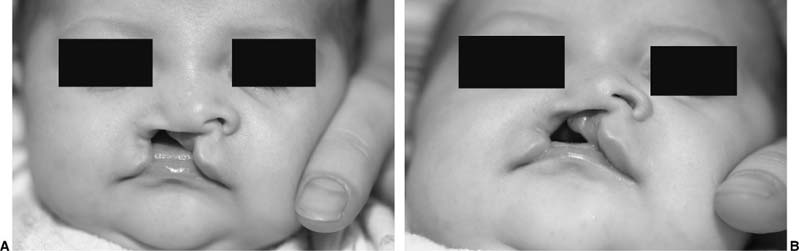
FIGURE 1-18 (A) A 6-week-old with a complete cleft of the right lip and palate. (B) Basal view of the same child with complete clefting of the lip and palate. Note the significant alar base asymmetry and the deviation of the columellar to the noncleft side.
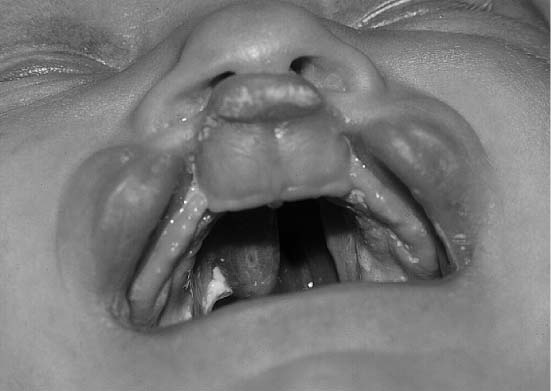
FIGURE 1-19 Basal view of a child with an incomplete bilateral cleft lip and palate. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.)
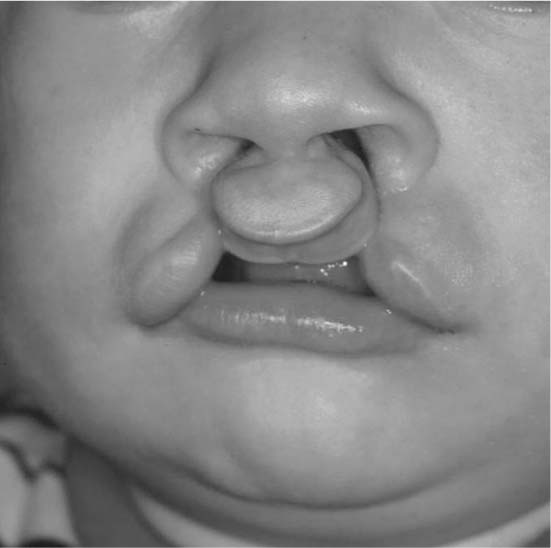
FIGURE 1-20 A 4-month-old boy with bilateral symmetric cleft lip and palate.
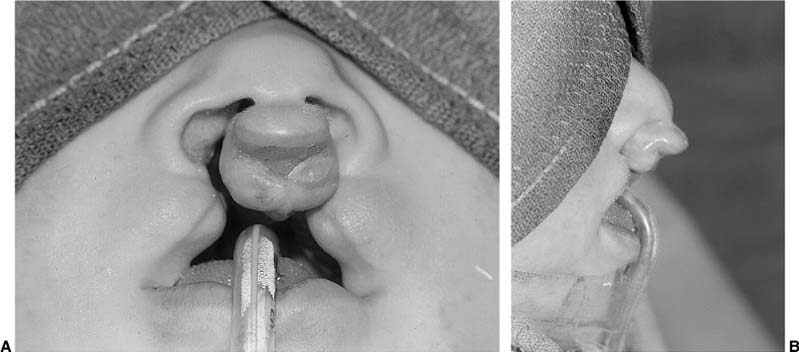
The unilateral or bilateral cleft lip deformity may be isolated or associated with clefts of the alveolus or palate. Because a greater interruption of lip development is required to create a bilateral cleft lip deformity, the bilateral lip deformity is more likely to be associated with clefting of the secondary palate than is the unilateral cleft lip deformity.
The primary palate is formed at between 4 and 8 week’s gestation simultaneously with the development of the upper lip and central maxillary alveolar ridge. The secondary palate is composed of the horizontal plate of the maxilla and palatine bone. The secondary palate fuses in the anterior-to-posterior direction at between weeks 8 and 12 embryologically. Interruption of the normal fusion of the secondary palate causes various degrees of palatal clefting.
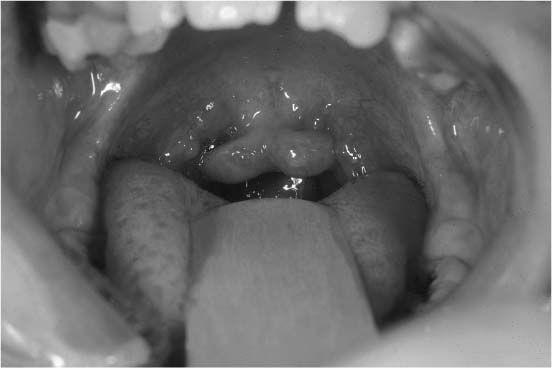
FIGURE 1-22 Intraoral view of a patient with an isolated bifid uvula.
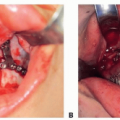
Clefts of the secondary palate can be variable in expression, depending on the timing and degree of interruption of palatal development. The most common condition and the smallest expression of clefting of the soft palate is the bifid uvula (Fig. 1-22). This common deformity occurs when there is a lack of normal fusion of the uvula. A submucous cleft of the soft palate occurs when there is dehiscence of the muscles of the soft palate (Fig. 1-23). In this condition, the palatal mucosa is intact, but there is incomplete development of the underlined palatal musculature. A submucous cleft palate often requires speech therapy and sometimes requires surgical repair. Full-thickness clefting of the soft palate can manifest with either an incomplete or complete secondary palatal cleft (Fig. 1-24). The incomplete secondary cleft occurs through the entire soft palate and into a portion of the hard palate. Complete clefting of the secondary palate may result from total interruption of the normal formation of the palate posterior to the incisive foramen.86
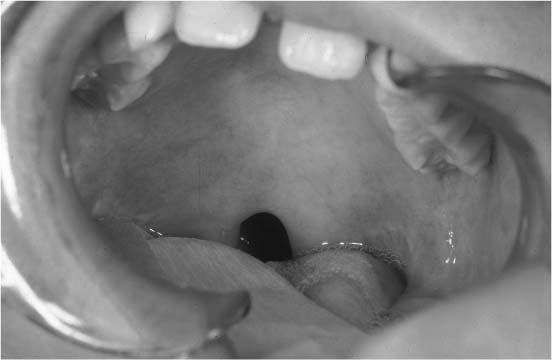
FIGURE 1-23 Intraoral view of a patient with a submucous cleft of the soft palate. Note the zona pellucida indicating the dehiscence of the soft palatal musculature.
Clefts of the secondary palate may be associated with complete cleft lips and are termed complete clefts of the lip and palate. Complete cleft lip and palate may be unilateral or bilateral. The complete unilateral cleft lip and palate usually involves attachment of the vomer to the maxillary palatal shelf of the noncleft side (Fig. 1-25). Complete bilateral clefts of the lip and palate usually have the central vomer and premaxilla detached from the two lateral palatal shelves (Fig. 1-26). In all cases, clefting of the lip and palate is variable in expression and typically follows known embryologic patterns.
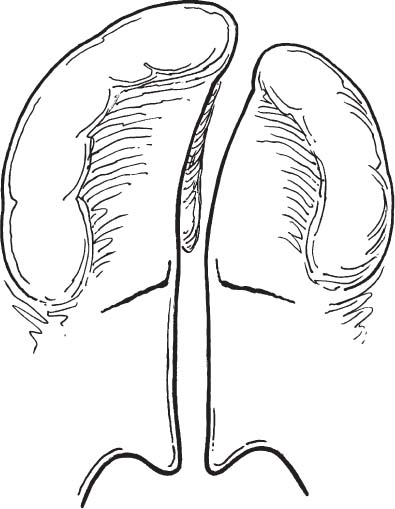
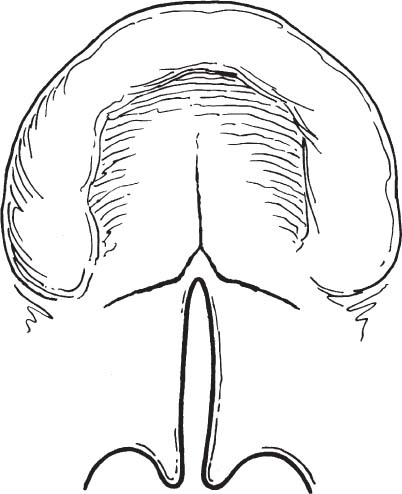
FIGURE 1-25 Schematic diagram of a complete unilateral cleft of the hard and soft palate. (From Papel ID. Facial Plastic and Reconstructive Surgery. 2nd Ed. New York: Thieme 2002. Used with permission.)
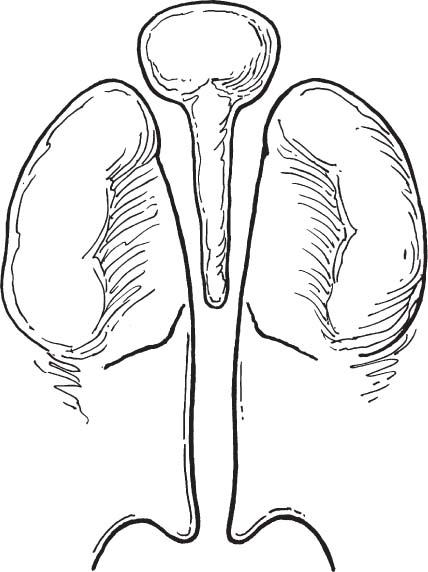
FIGURE 1-26 Schematic diagram of a complete bilateral cleft palate. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.)
TIMING AND PHILOSOPHY OF CLEFT REPAIR
The decision to repair a cleft lip or palate deformity is based on a variety of factors: speech development, facial growth, psychological impact on the child and family, and safety to undergo anesthesia. The timing of repair of various cleft deformities is outlined in Table 1-12.87 In patients with palatal clefting with or without clefts of the lip, tympanostomy tube placement is performed at age 3 to 6 months. This surgery is performed at an early age to (1) aerate the middle ear, (2) decrease conductive hearing loss, and (3) minimize the likelihood of chronic ear disease. In patients with cleft lip and palate, tympanostomy tube placement is performed at the same time as cleft lip repair. In these patients, surgery is performed at approximately 3 months of age and includes (1) cleft lip repair, (2) tympanostomy tube placement, and (3) closure of the floor of the nose and nasal rhinoplasty (when indicated). Cleft palate repair is performed prior to the initial development of speech. This is usually performed at 9 to 15 months of age. Long-acting tympanostomy tubes are placed at the time of palatoplasty.
| Cleft Procedure | Age at Repair |
|---|---|
| Cleft lip repair | 3 months |
| Tip rhinoplasty | |
| Tympanostomy tubes | |
| Palatoplasty | 9–18 months |
| T-tube placement | |
| Speech evaluation | 3–4 years |
| Velopharyngeal insufficiency workup and surgery (if necessary) | 4–6 years |
| Alveolar bone grafting | 9–11 years |
| Nasal reconstruction | 12–18 years |
| Orthoagnathic surgery (if necessary) | At completion of mandibular growth (>16 years) |
After cleft palate repair, patients are monitored carefully by the cleft and craniofacial team. Speech development is evaluated carefully beginning at 2 years of age. If velopharyngeal dysfunction (VPD) is identified, a VPD workup, including nasopharyngoscopy and video fluoroscopy, is performed. If the degree of VPD is significant, speech therapy is undertaken at approximately 3 years of age. If the patient does not respond to aggressive speech therapy, surgical treatment to correct the VPD is performed at between 4 and 6 years of age.
Other surgical treatments for cleft deformities include alveolar bone grafting at 9 to 11 years of age, internal and external nasal reconstruction (cleft septorhinoplasty) at 12 to 18 years of age, and orthognathic surgery after growth of the facial skeleton is complete. The need for these interventions is related to the initial deformity as well as the growth and development of the patient.
THE UNILATERAL CLEFT LIP DEFORMITY: PATHOLOGIC ANATOMY
A unilateral cleft of the upper lip involves alterations in all layers of the lip, including skin, muscle, mucosa, and underlying skeletal framework. The external form of the defect is determined by the extent of the underlying muscular and skeletal deformity. Subcutaneous (microform) cleft lip involves partial or total clefting of the upper lip musculature. Partial (incomplete) cleft lip involves skin, muscle, and mucosa, but may spare the underlying skeletal structures. Complete unilateral lip deformities involve all tissue layers.
The principal muscle of the lips is the orbicularis oris muscle (Fig. 1-27). The fibers of this muscle encircle the oral orifice within the substance of the lips.88 The orbicularis oris muscle consists anatomically of two parts: the superficial and the deep layers. This muscle is not a true sphincter, with the superficial and deep components arising as separate muscles from the modiolus at each oral commissure.89
In the unilateral cleft lip deformity, there is discontinuity of the orbicularis oris muscle in the region of the cleft (Fig. 1-28). The muscles of the unilateral cleft lip have two distinct differences when compared with normally developed lip musculature. First, the muscles are hypoplastic in the region of the cleft. Second, the muscles cannot cross the cleft gap and are prevented from attaching to their normal sites and have to find substitute insertions. Although it is obvious that no muscle crosses the cleft gap in complete lip clefting, the skin bridge in incomplete clefts has also been found to contain no functional musculature. Muscle dissections by Fara et al90 on stillborn babies with incomplete clefts confirm that the muscles in unilateral cleft lips are more hypoplastic on the medial side than on the lateral side of the cleft. Additionally, these dissections reveal that the upper lip muscles in incomplete clefts did not cross the cleft gap unless the skin bridge was at least one-third the height of the lip. Even if the orbicularis oris muscle is present in the skin bridging the incomplete cleft, the orientation of the musculature is abnormal.
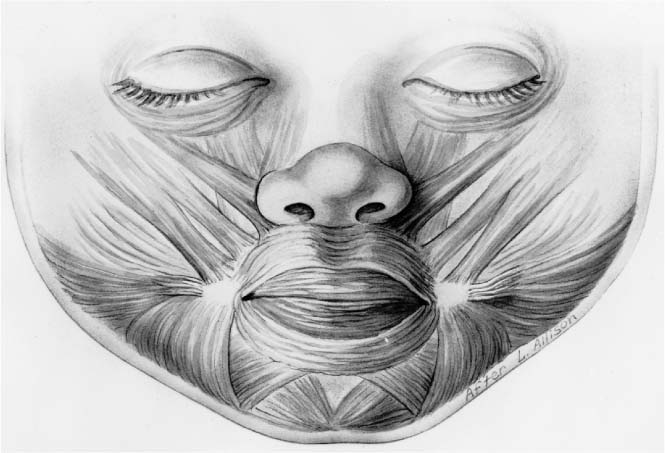
FIGURE 1-27 Schematic diagram of the orbicularis oris muscles as they relate to the other facial musculature. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.)
The major vascular supply to the lips and nose arises from the facial artery, which is a branch of the external carotid artery. The facial artery gives rise to the superior and inferior labial arteries at each oral commissure. The paired superior labial arteries anastomose in the midline of the upper lip, and the two inferior labial arteries similarly anastomose in the lower lip. In the unilateral cleft lip deformity, the aberrant vascular supply parallels the findings of the unilateral cleft lip musculature (Fig. 1-29). As with the musculature, the blood supply on the lateral aspect of the cleft is better developed than is the vasculature on the medial side. In the incomplete cleft lip deformity, a terminal branch of the superior labial artery crosses the epithelial skin bridge.
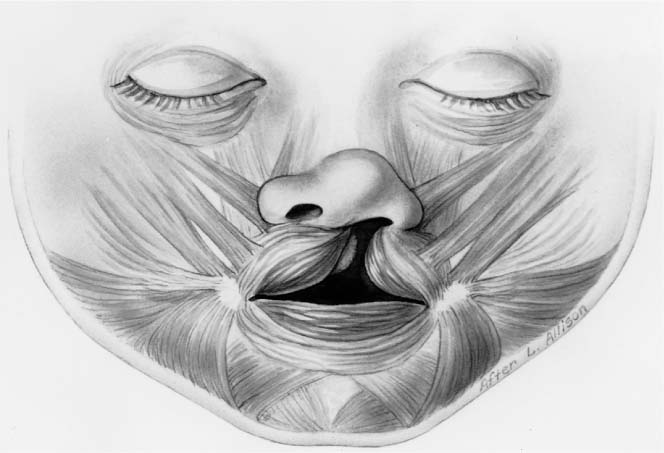
FIGURE 1-28 Schematic diagram of the orbicularis oris muscles associated with a unilateral cleft lip deformity. The muscles surrounding the upper lip cleft substitute with unnatural insertions along the cleft margin. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.)
UNILATERAL CLEFT LIP REPAIR
Many techniques have been described over the years for repair of the unilateral cleft lip deformity. Ambroise Paré91 repaired the unilateral cleft by freshening the cleft edges and skewering the two sides of the cleft with a long needle. The needle was then wrapped with thread in a figure-of-eight fashion. Rose92 and Thompson93 described modifications of straight-line closures for repair of the unilateral cleft lip deformity. All of the straight-line techniques close the cleft defect adequately, but often resulted in vertical scar contracture and notching of the upper lip. Wide complete unilateral cleft lips are also difficult to repair by the straight-line method. In the mid-20th century, various geometric closure techniques were proposed for repairing the unilateral cleft lip. Geometrical techniques, such as modified Z-plasties, quadrangular flaps, and triangular flaps, were designed to decrease the amount of lip shortening that occurred with cleft lip repair and to improve orbicularis oris muscle realignment and function.92,94,95 The triangular flap designed by Tennison and the quadrangular flap of Le Mesurier96 are reliable, consistent methods for decreasing vertical lip contraction in the unilateral cleft lip repair.
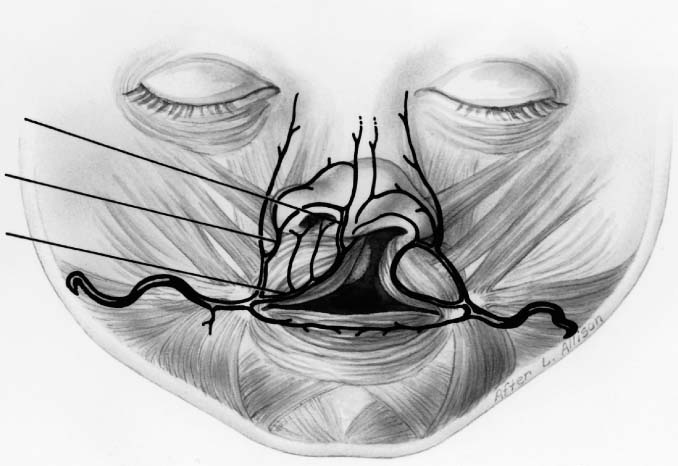
FIGURE 1-29 Schematic diagram of the aberrant vascular supply of the unilateral cleft lip. (From Papel ID. Facial Plastic and Reconstructive Surgery, 2nd ed. New York: Thieme, 2002, with permission.)
The primary advantage of geometric cleft lip repair techniques is that they provide a reproducible method to repair the lip. Exact measurements can be made with calipers to ensure reliable tension-free closure of the lip. The basic disadvantage of geometric designs is that the incisions always violate the curved philtral column on the noncleft side, creating a scar that crosses boundaries of known anatomic subunits. In addition, geometric repairs require exacting presurgical measurement and lack flexibility during surgical applications. In 1957, D. Ralph Millard97 incorporated aspects of multiple previously described repairs and developed the rotation-advancement flap technique for repair of the unilateral cleft lip deformity. This method maximizes flexibility for the surgery while minimizing the amount of normal lip tissue discarded. This method is the most commonly used technique today for unilateral cleft lip repair. The advantages and disadvantages of the Millard rotation-advancement flap technique are listed in Table 1-13.98
ROTATION-ADVANCEMENT REPAIR OF THE UNILATERAL CLEFT LIP DEFORMITY
The primary goals of unilateral cleft lip repair are to reconstruct normal lip anatomy and to restore lip function. Other goals include closure of the nasal floor, correction of nasal tip asymmetry and narrowing of the alveolar cleft.98 The rotation-advancement flap technique designs two major full-thickness flaps that can be approximated to repair the cleft without notching of the lip. This design allows reconstitution and reorientation of the orbicularis oris muscles.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


