The 22q11.2 deletion syndrome (22q11DS) may be associated with several palatal abnormalities, including overt cleft palate, submucosal cleft palate, palatopharyngeal disproportion, and velar hypotonia. The syndrome is the genetic disorder most commonly associated with velopharyngeal dysfunction (VPD). The complex causes of VPD in affected patients combine with the complexity of associated medical disorders to render surgical management of the velopharynx particularly challenging. Optimization of surgical outcomes requires precision in diagnosis, surgical management, and multidisciplinary care. This article provides an overview of 22q11DS and provides a review of the assessment and surgical management of VPD in affected individuals.
Key points
- •
The 22q11.2 deletion syndrome comprises the disorders also named DiGeorge syndrome and velocardiofacial syndrome.
- •
The 22q11.2 deletion syndrome is the syndrome most commonly associated with velopharyngeal dysfunction.
- •
Speech and language delays are common in patients with 22q11.2 deletion syndrome. The complexity of the associated speech and language disorders requires a multidisciplinary approach to management.
- •
Optimal surgical management of children with 22q11.2 deletion syndrome requires careful preoperative assessment, including imaging of the velopharyngeal mechanism.
- •
Functional and anatomic abnormalities of the velopharynx associated with 22q11.2 deletion syndrome demand that the surgical approach be tailored to each patient’s individual needs.
- •
Medical comorbidities associated with 22q11.2 deletion syndrome require rigorous preoperative assessment and postoperative monitoring.
Introduction and historical perspective
General Overview of 22q11.2 Deletion Syndrome
The 22q11.2 deletion syndrome (22q11DS) is a common genetic disorder typically involving cardiac defects, cognitive-behavioral problems, speech-language disorders, velopharyngeal dysfunction (VPD), and dysmorphic facial appearance. 22q11DS occurs in approximately 1 in 4000 births and is recognized as the most frequently occurring syndrome associated with VPD and palatal anomalies.
Over the past few decades, the nomenclature associated with this syndrome has varied significantly. In 1955, velofacial hypoplasia (referred to as Sedlackova syndrome) was first described by Sedlackova, a Czech phoniatrician, in a group of 26 children with congenital short soft palate, hypernasal speech, facial and ear dysmorphism, and muscle fiber abnormalities. In the late 1960s, Angelo DiGeorge identified a new condition termed the DiGeorge anomalad, which included hypocalcemia, hypoparathyroidism, immune deficiency, and cardiac defects. Later, in 1978, Shprintzen and colleagues reported on a syndrome with typical facies, including a prominent nose and retruded mandible, cardiovascular anomalies, cleft palate, and learning disabilities, later named velocardiofacial syndrome. It was later discovered that these overlapping phenotypes share a common underlying genomic disorder, 22q11DS, now known to be responsible for most cases with these previously described conditions.
Inheritance of the 22q11.2 deletion is autosomal dominant with incomplete penetrance and variable expressivity, and most cases are de novo. In 2005, it was reported that there was a bimodal distribution of age at diagnosis, with a peak at 6.5 years and remaining cases diagnosed in infancy (92% of which had a cardiac defect). For those children diagnosed after 2 years of age, most presented with a speech-language disorder and developmental delay. At present, the diagnosis of 22q11DS is based on a combination of clinical features and genetic confirmation of the deletion. The presence of the 22q11.2 deletion can be confirmed with fluorescence in situ hybridization (FISH) testing as well as microarray. With advancements in genetic testing, smaller and more specific mutations in the same region of the genome can also be identified (eg, TBX1 mutations).
More than 90% of children with 22q11DS present with speech and language disorders, VPD, and some degree of developmental delay. Other frequent clinical findings include cardiac defects and vascular anomalies, including medial displacement and tortuosity of the internal carotid arteries. Chiari malformation and cervical spine anomalies have also been observed. Upper respiratory illnesses and feeding difficulties are frequent in infancy. Typical craniofacial features ( Fig. 1 ) include a long midface and vertical maxillary excess, malar flatness, and mandibular retrusion. Prominent nose with a squared nasal root, bulbous nasal tip, and hypoplastic nasal alae are also consistent. Also reported are narrow palpebral fissures, long philtrum and thin upper lip, reduced facial affect, and minor external ear anomalies, such as dysmorphic helices. Endocrine, immune, and renal abnormalities are common. Behavioral difficulties, as well as significant psychiatric disorders, are also frequently identified. For a summary of additional phenotypic features, as well as general medical care guidelines for the syndrome, the reader is referred to Bassett and colleagues.
Speech and Language Disorders in 22q11DS
Generalized speech-language delays are commonly reported in 22q11DS, with many children showing early signs of velopharyngeal (VP) inadequacy. In addition to hypernasality, children may have articulation or phonological disorders, with a small percentage displaying motor speech disorders as well. Flat affect, reduced emotional expression, fast rate, high pitch, and monotone speech are also observed in many children. A significant proportion of preschool and school-aged children with 22q11DS show glottal stop substitutions, which are the most common compensatory error in the speech of children with cleft palate. The articulation skills of children with 22q11DS have been shown to be poorer than those of children with nonsyndromic cleft palate/VPD. A comprehensive review of information regarding speech-language disorders in 22q11DS is given by Gorlin and Baylis.
VPD in 22q11DS: Structural and Neuromuscular Considerations
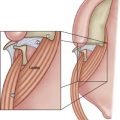
VPD in patients with 22q11DS has a complex causal origin ( Fig. 2 ). Although overt palate clefting is uncommon in 22q11DS, studies cite a high incidence of VPD and submucous clefting, as well as palatopharyngeal hypotonia, and obtuse cranial base or deep retropharynx (termed palatopharyngeal disproportion), all of which can compromise VP adequacy for speech. Ruotolo and colleagues reported that the increased pharyngeal depth associated with platybasia combines with increased pharyngeal width in affected patients to produce large increases in pharyngeal volume and VP valve area. Postadenoidectomy VPD is also common in children with this syndrome. Studies have reported an incidence of VPD in 22q11DS ranging from 64% to 87%, and, regardless of the cause, VPD and hypernasal speech are considered hallmark features of the syndrome. Children with 22q11DS tend to have hypernasal resonance of greater persistence, frequency, and severity than seen in other cleft and VPD populations. Thus, they pose a greater challenge to surgeons and speech pathologists.
In addition to structural abnormalities that contribute to VPD in 22q11DS, evidence has begun to surface regarding neuromuscular problems also affecting VP closure for speech. Pharyngeal muscle hypoplasia, cranial nerve abnormalities, and comorbid neuromotor speech disorders have also been reported as factors that could influence the degree of perceived hypernasality in speech, as well as negatively affect VP closure dynamics and surgical outcome.
Introduction and historical perspective
General Overview of 22q11.2 Deletion Syndrome
The 22q11.2 deletion syndrome (22q11DS) is a common genetic disorder typically involving cardiac defects, cognitive-behavioral problems, speech-language disorders, velopharyngeal dysfunction (VPD), and dysmorphic facial appearance. 22q11DS occurs in approximately 1 in 4000 births and is recognized as the most frequently occurring syndrome associated with VPD and palatal anomalies.
Over the past few decades, the nomenclature associated with this syndrome has varied significantly. In 1955, velofacial hypoplasia (referred to as Sedlackova syndrome) was first described by Sedlackova, a Czech phoniatrician, in a group of 26 children with congenital short soft palate, hypernasal speech, facial and ear dysmorphism, and muscle fiber abnormalities. In the late 1960s, Angelo DiGeorge identified a new condition termed the DiGeorge anomalad, which included hypocalcemia, hypoparathyroidism, immune deficiency, and cardiac defects. Later, in 1978, Shprintzen and colleagues reported on a syndrome with typical facies, including a prominent nose and retruded mandible, cardiovascular anomalies, cleft palate, and learning disabilities, later named velocardiofacial syndrome. It was later discovered that these overlapping phenotypes share a common underlying genomic disorder, 22q11DS, now known to be responsible for most cases with these previously described conditions.
Inheritance of the 22q11.2 deletion is autosomal dominant with incomplete penetrance and variable expressivity, and most cases are de novo. In 2005, it was reported that there was a bimodal distribution of age at diagnosis, with a peak at 6.5 years and remaining cases diagnosed in infancy (92% of which had a cardiac defect). For those children diagnosed after 2 years of age, most presented with a speech-language disorder and developmental delay. At present, the diagnosis of 22q11DS is based on a combination of clinical features and genetic confirmation of the deletion. The presence of the 22q11.2 deletion can be confirmed with fluorescence in situ hybridization (FISH) testing as well as microarray. With advancements in genetic testing, smaller and more specific mutations in the same region of the genome can also be identified (eg, TBX1 mutations).
More than 90% of children with 22q11DS present with speech and language disorders, VPD, and some degree of developmental delay. Other frequent clinical findings include cardiac defects and vascular anomalies, including medial displacement and tortuosity of the internal carotid arteries. Chiari malformation and cervical spine anomalies have also been observed. Upper respiratory illnesses and feeding difficulties are frequent in infancy. Typical craniofacial features ( Fig. 1 ) include a long midface and vertical maxillary excess, malar flatness, and mandibular retrusion. Prominent nose with a squared nasal root, bulbous nasal tip, and hypoplastic nasal alae are also consistent. Also reported are narrow palpebral fissures, long philtrum and thin upper lip, reduced facial affect, and minor external ear anomalies, such as dysmorphic helices. Endocrine, immune, and renal abnormalities are common. Behavioral difficulties, as well as significant psychiatric disorders, are also frequently identified. For a summary of additional phenotypic features, as well as general medical care guidelines for the syndrome, the reader is referred to Bassett and colleagues.
Speech and Language Disorders in 22q11DS
Generalized speech-language delays are commonly reported in 22q11DS, with many children showing early signs of velopharyngeal (VP) inadequacy. In addition to hypernasality, children may have articulation or phonological disorders, with a small percentage displaying motor speech disorders as well. Flat affect, reduced emotional expression, fast rate, high pitch, and monotone speech are also observed in many children. A significant proportion of preschool and school-aged children with 22q11DS show glottal stop substitutions, which are the most common compensatory error in the speech of children with cleft palate. The articulation skills of children with 22q11DS have been shown to be poorer than those of children with nonsyndromic cleft palate/VPD. A comprehensive review of information regarding speech-language disorders in 22q11DS is given by Gorlin and Baylis.
VPD in 22q11DS: Structural and Neuromuscular Considerations
VPD in patients with 22q11DS has a complex causal origin ( Fig. 2 ). Although overt palate clefting is uncommon in 22q11DS, studies cite a high incidence of VPD and submucous clefting, as well as palatopharyngeal hypotonia, and obtuse cranial base or deep retropharynx (termed palatopharyngeal disproportion), all of which can compromise VP adequacy for speech. Ruotolo and colleagues reported that the increased pharyngeal depth associated with platybasia combines with increased pharyngeal width in affected patients to produce large increases in pharyngeal volume and VP valve area. Postadenoidectomy VPD is also common in children with this syndrome. Studies have reported an incidence of VPD in 22q11DS ranging from 64% to 87%, and, regardless of the cause, VPD and hypernasal speech are considered hallmark features of the syndrome. Children with 22q11DS tend to have hypernasal resonance of greater persistence, frequency, and severity than seen in other cleft and VPD populations. Thus, they pose a greater challenge to surgeons and speech pathologists.
In addition to structural abnormalities that contribute to VPD in 22q11DS, evidence has begun to surface regarding neuromuscular problems also affecting VP closure for speech. Pharyngeal muscle hypoplasia, cranial nerve abnormalities, and comorbid neuromotor speech disorders have also been reported as factors that could influence the degree of perceived hypernasality in speech, as well as negatively affect VP closure dynamics and surgical outcome.
Patient assessment
Speech Assessment: Perceptual Evaluation and Instrumental Assessment
In general, the protocol for assessment of speech in patients with 22q11DS is similar to that encountered in other populations with cleft palate or VPD. For the purposes of this article, syndrome-specific considerations are highlighted. All patients with 22q11DS should be followed by a cleft/craniofacial team that is familiar with management of VPD. The speech-language pathologist should ideally begin following the child starting in infancy, monitoring speech and language acquisition and managing therapy needs. For toddlers and preschoolers, the focus of speech assessment and treatment is articulation and language. The speech pathologist should closely monitor the child’s articulation for the presence of compensatory articulation errors (eg, glottal stops), and, if present, speech therapy should aggressively target their remediation as soon as they are identified. The child needs a phonetic inventory that includes at least 1 or 2 oral pressure consonants (eg, P, B, T) with correct placement in order to participate in VP imaging during speech. In general, most children with 22q11DS are able to produce an adequate speech sample that allows perceptual and instrumental assessment of VP function by 4 to 5 years of age. The speech evaluation should include conversational speech, repetition of standard sentences, and articulation testing. An intraoral examination should always be completed and additional perceptual assessment techniques (eg, nasal mirror testing, nasal occlusion) should also be considered. When symptoms of VPD are observed, instrumental assessment may be helpful as an adjunct to the perceptual evaluation. This assessment may include use of the Nasometer (KayPENTAX), an acoustic measurement that correlates with resonance, and/or pressure-flow testing (eg, PERCI-SAR, Microtronics), which allows measurement of intraoral pressure, nasal airflow, and estimated VP orifice size. Nasopharyngoscopy and/or multiview videofluoroscopy during speech should then be completed to provide additional information on VP function during speech and assist with surgical planning.
Imaging Recommendations
Regardless of which imaging modality (or modalities) is selected, the quality of the VP imaging study is directly related to the accuracy of the speech sample produced. Only patients who are able to produce at least a minimally accurate speech sample (in terms of oral consonant production) should undergo imaging, in order to obtain the most accurate information about VP function. If children with severe compensatory articulation disorders (eg, only produce glottal stops during the speech sample) undergo imaging, the outcome of the procedure is known without even conducting the study: lack of VP closure characterized by a large gap and minimal to no VP motion. Instead, if the child is able to produce at least 1 or 2 plosive consonants (ideally P, B, T, D), even if they are weak in pressure or accompanied by nasal emission, the examiner is able to see the patient’s true attempts at maximum VP closure during speech. This assessment also allows the most accurate surgical planning possible, treating only the VP gap that remains during the accurate speech attempts, and provides a more accurate characterization of movement of the VP mechanism. A well-trained craniofacial speech pathologist can determine what speech sample is most appropriate to use in the study, based on the child’s phonetic inventory, articulation accuracy, and desired complexity of the sample (eg, oral-only words vs phrases or sentences). In terms of the type of imaging procedure, multiview videofluoroscopy and nasopharyngoscopy both offer distinct advantages, as well as disadvantages, however, these are beyond the scope of discussion of this chapter.
Timing of VPD Surgery
Children with 22q11DS traditionally undergo VPD surgery at slightly later ages than their peers with cleft palate or nonsyndromic VPD. On average, surgical intervention is usually recommended at approximately 4 to 6 years of age or older. Treatment is later because of a variety of factors including, but not limited to, (1) diagnosis of VPD is often later in 22q11DS than that for children born with overt cleft palate, because children born with overt structural anomalies tend to be monitored for VPD by their craniofacial team from infancy; (2) other emergent medical issues (eg, cardiac, airway) in children with 22q11DS may take precedent over VP management; (3) many children with 22q11DS have severe articulation disorders that require a longer duration of speech therapy to correct until the diagnosis of VPD is confirmed and VP imaging can be completed successfully (ie, sufficiently accurate articulation and adequate cooperation for the study).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


