Fig. 6.1
Scheme of a cornified layer barrier: the lamella granules and the keratohyalin granules in the epidermis play important roles in the formation of a mature stratum corneum. The corneocytes are surrounded by a cornified cell envelope, and the keratin fiber is bundled with filaggrin. Filaggrin is degraded into a natural moisturizing factor. A firm barrier is formed via adherence between corneocytes and corneodesmosomes. Serine proteases break down the corneodesmosome, and old corneocytes will be detached
6.3 Genetic Abnormalities in Barrier Dysfunction
The identification of genetic abnormalities in expression of serine proteases, serine protease inhibitors, or filaggrin in the stratum corneum has promoted the understanding of the hereditary predisposition of atopic dermatitis. The mechanisms of barrier dysfunction found in Netherton syndrome, which shows excess activity of serine protease, provided us with useful information for understanding the factors that influence atopic predisposition.
In Netherton syndrome, excess activation of serine protease, resulting from a loss-of-function mutation Glu420Lys (single-nucleotide polymorphism [SNP]) in the serine protease inhibitor Kazal type 5 (SPINK5) (disease specificity of this mutation remains questionable), causes an excessive activation of serine proteases in the stratum corneum and leads to extravagant degradation of both corneodesmosome and lipid processing enzymes [8, 9] (Fig. 6.1). Aberrant degradation of these molecules involved in stabilization of the stratum corneum leads to development of skin phenotypes similar to atopic dermatitis, which is characterized by fragile stratum corneum with abnormally increased permeability [8].
Netherton syndrome is considered a disease that should be excluded from atopic dermatitis according to criteria for atopic dermatitis created by the Japanese Dermatological Association [10]. Additionally accelerated activation of serine protease inhibits secretion of lamella granules via activation of plasminogen activator type 2 receptor (PAR2), leading to barrier dysfunction [11].
Evidence of the close relationship between the etiology of atopic dermatitis and the loss-of-function mutation in filament-aggregating protein (filaggrin [FLG]) suggests that both structural and functional abnormalities of the stratum corneum may be the basis of the pathogenic etiology of atopic dermatitis [12]. FLG is a component of F-type keratohyalin granules, and its loss-of-function mutation has also been confirmed in atopic dermatitis patients as well as in ichthyosis vulgaris patients, and about 2/3 of the ichthyosis vulgaris cases were complicated with allergic diseases, such as atopic dermatitis, allergic rhinitis, and asthma [1]. Common physiological response in keratinization results in dephosphorylation and degradation of the FLG precursor, producing the FLG monomer, which agglutinates keratin in corneocytes to be irrefrangible. Further degradation of the FLG monomer contributes to moisture retention by the stratum corneum as a natural moisturizing factor. Although decreased FLG impairs the water-retention capacity of the stratum corneum, the minimum requirement of FLG for preventing skin dryness has yet to be defined. Expression levels of FLG are affected by environmental factors and skin inflammation [13]. Furthermore, the mutation of FLG has been confirmed only in some cases with atopic dermatitis. A recent study of Japanese subjects found that the prevalence of FLG mutations is 11.1% (n = 820) [14]. Thus, we should keep in mind that decreased FLG could be both a cause and a consequence of the skin conditions in atopic dermatitis.
6.4 Epidermal Barrier: Role of Tight Junctions
The epidermis should be the final sophisticated defense against pathogens (Fig. 6.2). To avoid entry of pathogens into the body and to avoid water leakage through paracellular space between the epidermal cells, cell adhesion structures called “tight junctions” exist. Tight junctions in the epidermal granular layer have been confirmed by both electron microscopy and immunofluorescence, and these tight junctions form the barrier of the paracellular space to regulate permeability [15]. On another front, Langerhans cells exist inside the epidermis and elongate their dendrites to reach the outside of tight junction to sample pathogens [16]. Even in this case, tight junctions are formed between extended dendrites of activated Langerhans cells and epidermal granular cells [16]. This finding shows the importance of epidermal tight junctions. Every tight junction consists of transmembrane proteins (e.g., claudins, occludin, and tricellulin), cytoplasmic adaptor proteins, and cytoskeletal linkers (e.g., zonula occludens (ZO) proteins, etc.) [15, 17, 18] (Fig. 6.2). Claudins, which are essential tight junction transmembrane proteins, have 27 family members [15]. Claudin-1 and claudin-4 are expressed in skin epithelial cells and have roles in paracellular barrier function and possibly regulation of cell differentiation, respectively [15]. Claudin-1 is the major component of the epidermal tight junction and is indispensable to prevent fatal dehydration, as shown by claudin-1 knockout (Cldn1KO) animals [15, 17, 18]. An SNP in the claudin-1 (Cldn1) gene and inflammation-mediated decreased expression of claudin-1 have been observed in subjects with atopic dermatitis and have been found to be involved in the etiology of atopic dermatitis [19, 20]. More recently, claudin-1 was found to regulate the adequacy of the paracellular barrier in a dose-dependent manner, and decreased claudin-1 was found to cause skin manifestations resembling those in atopic dermatitis [21].
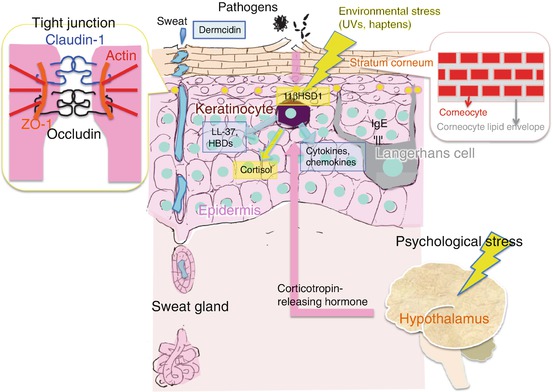
Fig. 6.2
Scheme of the defense mechanisms of the skin. Pathogens penetrate from the outside of the skin via a damaged stratum corneum. Pathogens or substances derived from pathogens pass through the tight junction, activating keratinocytes via pattern-recognition receptors. The activated keratinocytes release antimicrobial peptides (e.g., LL-37, human β-defensins [hBDs]), cytokines, and/or chemokines to recruit inflammatory cells. Keratinocytes exposed to environmental stresses will activate 11βHSD1 and produce cortisol to reduce the negative impact of environmental stimuli. Psychological stresses activate the hypothalamus in the central nervous system, causing release of corticotropin-releasing hormone (CRH) to regulate epidermal homeostasis
Fatal dehydration in Cldn1KO mice has been a barrier to investigation of the pathogenic involvement of claudin-1 in atopic dermatitis. Thus, Tokumasu et al. generated six types of Cldn1 knockdown (Cldn1KD) mice with different expression levels of Cldn1 and found dysfunction of the epidermal barrier caused by decreased Cldn-1 levels to less than half [21]. Decreased expression of Cldn-1 affected differentiation of keratinocytes and increased the number of K5-positive or ectopically proliferating suprabasal cells [21].
Cldn1KD mice develop age-related skin manifestations. Cldn1KD mice and moderately decreased Cldn1 (Cldn1 ∆/∆) mice develop wrinkled skin and dry hair at around 1 week and 2 weeks of age, respectively. This skin phenotype disappeared by 8 weeks of age. This age-related phenotypic change appears similar to the clinical course of human pediatric atopic dermatitis cases, as most human pediatric patients outgrow the disease. Cldn1KD mice with severely decreased Cldn1 (Cldn1 ∆/−) show more severe skin manifestations with severe desquamation and wrinkled skin at the age of 8 weeks or older [21]. These findings indicate that expression of Cldn-1 will affect the severity of atopic dermatitis symptoms. Also, increased numbers of skin-homing innate immune cells, such as neutrophils and macrophages, in Cldn1KD mice indicated that epidermal barrier dysfunction requires functional augmentation of innate immunity to cope with penetration of certain pathogens from the outside [21].
Further elucidation of the precise mechanism of epidermal barrier dysfunction may lead to a better understanding of the pathogenesis and natural clinical course of atopic dermatitis. The epidermal barrier may, therefore, provide a therapeutic target and may contribute to formulation of novel therapeutic strategies for management of atopic dermatitis.
6.5 Defense Mechanism of Skin in Atopic Dermatitis
Host defense mechanisms, such as regulating skin permeability (as mentioned above) and innate immunity, are also impaired in atopic dermatitis. These abnormalities lead to the acquisition of an accompanying bacterial infection (e.g., impetigo contagiosa) in atopic dermatitis patients. It has been confirmed that the frequency of the colonization of Staphylococcus aureus is different between lesional and non-lesional skin and is increased in lesional skin [22]. Viral infections (e.g., Kaposi’s varicelliform eruption and molluscum contagiosum) and fungal infections (e.g., tinea corporis and colonization of Malassezia) are also frequently found in large areas of the body of subjects with atopic dermatitis [23]. Aberrantly increased permeability of skin and/or dysfunction of innate immune responses are involved in the increased susceptibility to infection in atopic dermatitis [23] (Fig. 6.2). The innate immune system in the skin surface is formulated with genetically encoded molecules that are inherent in the human skin and skin appendages themselves [23]. Receptors that recognize the highly conserved structure in pathogens, the so-called pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs) and collectin proteins, are expressed on keratinocytes and Langerhans cells and induce the innate immune response by binding with pathogen-associated molecular patterns (PAMPs) characterized by cell wall products such as LPS, peptidoglycan, and viral double-stranded RNA derived from common pathogens [23, 24]. The innate immune response is characterized by the release of antimicrobial peptides (AMPs), chemokines, and cytokines and will immediately inhibit the infection [23, 25]. AMPs are released from keratinocytes and are also contained in sweat [23, 26] (Fig. 6.2). Continuous secretion of dermcidin from sweat glands contributes to the regulation of the proper skin surface microbiome [27].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


