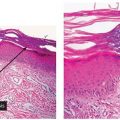
Figure 27-1 Post-inflammatory hyperpigmentation. There are numerous melanophages in the papillary dermis along with a mild perivascular lymphocytic infiltrate. The epidermis is normal.
Pathogenesis. Direct stimulation of melanocytes by inflammatory mediators such as IL-1, endothelin 1, and stem cell factor (56) as well as fibroblast-derived melanogenic growth factors have been proposed to play a role in post-inflammatory hyperpigmentation (57).
Differential Diagnosis. Vitiligo, idiopathic guttate hypomelanosis, progressive macular hypopigmentation, infections (tinea versicolor, leprosy, onchocerchiasis, pinta), nevus depigmentosus, drug-induced hyperpigmentation, systemic diseases (Addison disease, hemochromatosis, hyperthyroidism, renal and hepatic failure, systemic lupus erythematosus, dermatomyositis), nutritional diseases (pellagra, malabsorption), onchronosis, lymphoma.
Principles of Management. Treatment of the underlying cause. In addition, for hyperpigmentation: bleaching creams containing hydroquinones, retinoids, topical corticosteroids, chemical peels, azelaic acid, laser treatment, and camouflage (58,59). For hypopigmentation: topical corticosteroids, topical calcineurin inhibitors, phototherapy, skin grafts (60).
BERLOQUE DERMATITIS AND PHYTOPHOTODERMATITIS
Clinical Summary. Localized hyperpigmented patches produced by furocoumarin-containing oil of Bergamot found in perfumes have a distinctive clinical presentation. The patches assume drop like shapes resembling pendants (French: berloque) and are usually located on the sides of the neck or retroauricular areas. Contact with plants containing furocoumarins may also produce hyperpigmented patches and include lime, wild and cow parsnip, wild carrot, bergamot orange, and fig (61).
Histopathology. Features identical to post-inflammatory hyperpigmentation are found, including increased epidermal melanin and melanophages in the superficial dermis. Variable chronic inflammation is present.
Differential Diagnosis. Post-inflammatory hyperpigmen-tation.
Principles of Management. Avoidance of offending agents, topical corticosteroids, bleaching agents.
CHEMICAL DEPIGMENTATION
Chemical leukoderma is an acquired depigmentation resembling vitiligo caused by repeated exposure to agents that destroy melanocytes in genetically susceptible individuals. Hydroquinones, phenols, catechols as well as sulfhydryl compounds are the most common causes and are found in bleaching agents, adhesives, cosmetics, hair dyes, photographic processing materials, fabrics treated with azo dyes, and antioxidants used in the manufacture of rubber (62–65). Depigmentation may also occur in body sites remote from chemical contact, presumably from systemic absorption or inhalation. Systemic medications (fluphenazine, chloroquine, and imatinib mesylate), optic medications and compounds containing mercury, arsenic, and cinnamic aldehyde have also been reported to cause depigmentation.
Histopathology. Features indistinguishable from vitiligo are found (62). Decreased or absent melanocytes are present with a variable superficial perivascular lymphocytic infiltrate.
Differential Diagnosis. Vitiligo, infections (tinea versicolor, leprosy, onchocerchiasis, pinta), genetic syndromes (piebaldism, hypomelanosis of Ito, tuberous sclerosis, Waardernburg syndrome, Vogt–Koyanagi–Harada syndrome), post-inflammatory hypopigmentation, nevus depigmentosus, idiopathic guttate hypomelanosis, progressive macular hypomelanosis.
Principles of Management. Avoidance of offending agents, treatments used for vitiligo (see vitiligo section).
IDIOPATHIC GUTTATE HYPOMELANOSIS
Clinical Summary. Idiopathic guttate hypomelanosis is a commonly acquired benign disorder of unknown etiology that produces few to numerous asymptomatic sharply circumscribed, 2- to 6-mm white macules. These lesions occur predominantly on the legs and forearms of persons over 30 years of age (66,67).
Histopathology. Fontana–Masson stain of lesional skin shows decreased melanin contact compared to perilesional skin. The lesional borders are sharply demarcated. There is melanocyte degeneration and decreased melanosomes within the hypomelanotic epidermis. The melanin granules are irregularly distributed, and there is a significant reduction in the number of melanocytes (68–70).
Pathogenesis. On electron microscopic examination, the scattered residual melanocytes in lesional skin are round and less dendritic with fewer melanosomes that are incompletely melanized (71). Chronic sun exposure, trauma, autoimmunity, and genetic factors like HLA-DQ3 have been proposed to contribute to the pathogenesis of this disease.
Differential Diagnosis. Post-inflammatory hypopigmentation, progressive macular hypopigmentation, pityriasis versicolor, vitiligo.
Principles of Management. Topical calcineurin inhibitors (72), dermabrasion, cryotherapy (73), lasers (74).
PROGRESSIVE MACULAR HYPOPIGMENTATION
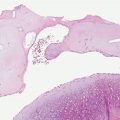
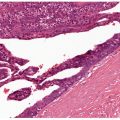
Clinical Summary. Progressive macular hypopigmentation is a disorder of unclear etiology that is characterized by ill-defined, asymptomatic hypopigmented macules on the central trunk, sometimes extending to the neck and face predominantly in young adults (Fig. 27-2) (75). The lesions may be discrete or eventually become confluent and occur without previous inflammation or skin disease (76). Complete regression has been reported (77).
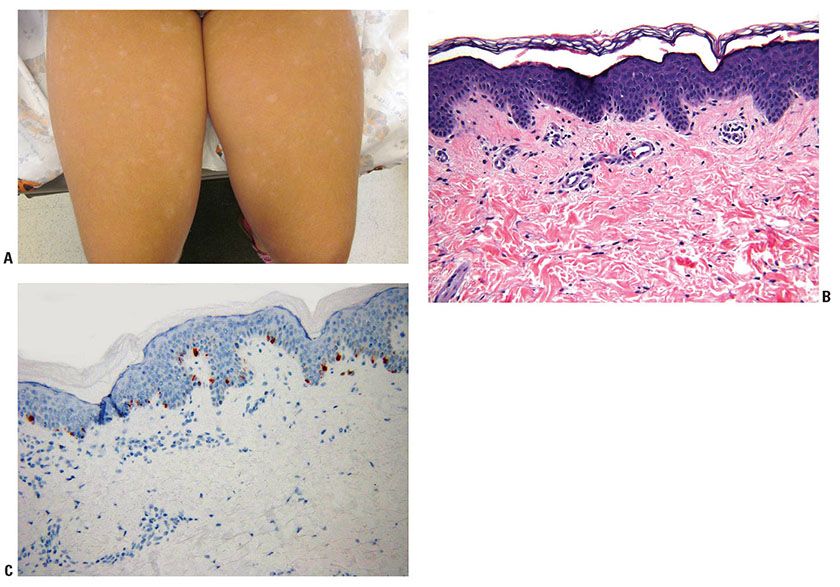
Figure 27-2 Progressive macular hypopigmentation. A: Ill-defined hypopigmented macules. B: Normal-appearing epidermis and dermis. C: Melan-A staining showing normal number of junctional melanocytes.
Histopathology. Histologic features of lesional and nonlesional skin may be indistinguishable (Fig. 27-2). A subtle decrease in melanin content may be present in lesional skin, and staining with Fontana–Masson may demonstrate reduction in melanin within the basal cell layer (76,78). Electron microscopy studies found less dense pigmentation and fewer mature melanosomes in lesional skin compared with normal skin (79,80).
Pathogenesis. Different subtypes of Propionibacterium acnes have been suggested to play a role (81). Propionibacterium acnes has been cultured and identified histologically in follicles of lesional skin but not follicles or interfollicular areas of healthy appearing adjacent skin. It has been proposed that the bacteria may interfere with melanogenesis in these skin lesions (78).
Differential Diagnosis. Idiopathic guttate hypomelanosis, post-inflammatory hypopigmentation, pityriasis versicolor, vitiligo.
Principles of Management. Phototherapy (82), benzoyl peroxide, clindamycin (83).
PSORALEN AND ULTRAVIOLET A–INDUCED PIGMENT ALTERATIONS
Clinical Summary. Psoralen and ultraviolet A (PUVA) therapy consists of the oral ingestion or topical application of a furocoumarin-containing psoralen compound followed by patient exposure to high-intensity UVA radiation in a controlled-light-box setting. It is widely used for treating psoriasis and cutaneous T-cell lymphoma and less commonly used for a variety of other dermatoses including repigmentation therapy of vitiligo.
A spectrum of clinical and histologic changes is seen during and after PUVA therapy. Acutely, the phototoxic erythema peaks at 48 to 72 hours. Hyperpigmentation (tanning) is delayed for several days and more pronounced and longer lasting than that induced by ultraviolet B (UVB) exposure (84). Prolonged therapy leads to photoaging with cutaneous atrophy, fine wrinkling, mottled hyperpigmentation and hypopigmentation, telangiectasias, and loss of elasticity (85). Stellate lentigines or PUVA freckles often develop after prolonged therapy (86). Predisposing factors for the appearance of PUVA lentigines include skin type, number of treatments and sex (males more than females). There is also a dose-dependent increase in the risk of melanoma and squamous cell carcinoma (87), especially in the genital areas (88).
Histopathology. The histologic changes of PUVA-induced acute phototoxicity are similar to sunburn, with numerous pink apoptotic cells (“sunburn cells”) scattered throughout the epidermis. Early in the therapeutic course, the melanocytes in the basalar epidermis and keratinocytes in the mid-spinous layer are heavily melanized. Prolonged therapy leads to gradual flattening of the rete ridges, telangiectasias, increased papillary dermal acid mucopolysaccharide deposition, thinning and basophilic degeneration of elastic fibers, and hyperplasia and fragmentation of elastic fibers (89). Hyperpigmented skin without other clinical changes frequently demonstrates small foci of keratinocytic nuclear atypia and loss of the normal maturation pattern (90). Actinic keratoses, Bowen disease, squamous cell carcinomas and melanomas may develop. PUVA freckles or lentigines show increased numbers of melanocytes and elongation of the rete ridges with focal atypical melanocytes (91).
Pathogenesis. On electron microscopy, melanosomes are diffusely distributed throughout the epidermis (92). There is basement membrane thickening with focal dissolution. Elastic fibers in the dermis appear homogenized and fragmented (93). In the deep dermis, there is reduplication of the basal layer of capillaries and increased pinocytosis of endothelial cells (94).
Differential Diagnosis. For PUVA lentigines includes lentigo simplex, solar lentigo, and pigmented actinic keratosis.
Principles of Management. Discontinuation of PUVA therapy.
DRUG-INDUCED PIGMENTARY ALTERATIONS
Clinical Summary. Drug-induced pigmentary alterations are quite common. Implicated agents include nonsteroidal anti-inflammatory drugs, antimalarials, tetracyclines, psychotropics including phenothiazines, antineoplastic agents including cytotoxics, heavy metals including gold, amiodarone, amitriptyline, imipramine, and clofazimine (95,96). A variety of pathogenic mechanisms are seen, including increased production of melanin leading to hyperpigmentation of basalar keratinocytes, vacuolar change at the dermal–epidermal junction with pigmentary incontinence and dermal melanophages, binding of the drug to melanin and deposition of stable complexes, and deposition of the drug itself as granules in the dermal extracellular matrix or in macrophages. In addition, drugs may induce the synthesis of special pigments, such as lipofuscin, or damage blood vessels leading to red blood cell extravasation and hemosiderophages (95,96). Clinical features vary from generalized hyperpigmentation to specific patterns attributed to individual drugs such as the flagellated streaks attributed to bleomycin (97).
Histopathology. Minocycline may cause three patterns of hyperpigmentation (98). The first pattern is a blue-black pigmentation of acne and other scars due to black pigment deposition in macrophages and free in the dermis. The second pattern consists of gray-blue pigmentation of the anterior legs and arms due to deposition of drug complexes and protein extracellularly in the dermis and commonly involves the eccrine ducts and other adnexa. The third pattern consists of brown discoloration of sun-exposed sites due to increased basalar keratinocytic melanin.
Antimalarial agents deposit yellow to dark-brown pigment granules in dermal macrophages and extracellularly (95). Both chloroquine and hydroxychloroquine bind melanin (99). Phenothiazines commonly deposit electron-dense granules in dermal macrophages. The granules are composed of a drug metabolite and melanin leading to progressive blue pigmentation on sun-exposed skin (100). Amiodarone may provoke a photoallergic reaction with blue-gray pigmentation due to the deposition of lipofuscin granules in macrophages around superficial dermal blood vessels. These are accentuated by periodic acid–Schiff (PAS) staining. Lipofuscinosis may also be caused by antibiotics (95). Imipramine deposits dermal round golden-brown bodies free in the dermis and within macrophages, which may represent abnormal drug metabolite–melanin complexes (101). Electron microscopy shows electron-rich granules within lysosomes and free in the dermis rich in copper and sulfur (102).
Chemotherapy agents commonly produce both generalized and localized hyperpigmentation of the basalar keratinocytes. Bleomycin, busulfan, doxorubicin, daunorubicin, fluorouracil, cyclophosphamide, and carmustine additionally may demonstrate melanophages in the superficial dermis (97). Globules of mercury have been found in dermal and subcutaneous abscesses after oral ingestion (103).
Differential Diagnosis. Post-inflammatory hyperpigmentation, phototoxic hyperpigmentation, systemic diseases (Addison disease, hemochromatosis, hyperthyroidism, renal and hepatic failure, systemic lupus erythematosus, dermatomyositis), nutritional diseases (pellagra, malabsorption), ochronosis, lymphoma.
Principles of Management. Discontinuation of the offending drug, sun protection, laser therapy.
PIGMENTARY PRESENTATIONS OF CUTANEOUS AND SYSTEMIC DISEASES
Clinical Summary. A large number of cutaneous and systemic diseases present with changes in pigmentation. These diseases can be nutritional, autoimmune, metabolic, endocrine, neoplastic, or infectious in etiology. Some have localized versus generalized patterns of pigment change. Mycosis fungoides may present with hypopigmented (104,105) or hyperpigmented (106) lesions. The former occurs in children and young adults, is slowly progressive, and often is present for many years before diagnosed. Sarcoidosis (107) may display hypopigmented skin lesions but usually demonstrates some degree of clinical induration. Darier (108) disease may also present with hypopigmentation. Although pigmented variants of basal cell carcinoma are not uncommon, pigmented Bowen disease (109), pigmented Paget disease (110), and depigmented extramammary Paget disease (111) can be difficult to recognize clinically. Skin metastases from breast carcinoma may be pigmented and can even simulate malignant melanoma (112). As well, generalized hyperpigmentation may be the presenting sign of metastatic malignant melanoma and primary pituitary tumors.
Histopathology. The histologic changes are those of the primary dermatosis. In hypopigmented mycosis fungoides, variable melanin pigmentary incontinence occurs (105), whereas hyperpigmented mycosis fungoides shows striking melanin pigment in the basal layer and giant melanin pigment granules throughout the spinous layer (106). No alteration of melanocytes is present in hypopigmented sarcoidosis, and no appreciable change in melanin pigment is noted (107). Decreased melanin pigment is seen in the basal layer of hypopigmented Darier disease (108). Pigmented Bowen disease shows abundant melanin pigment in basal keratinocytes, highly dendritic melanocytes, and numerous melanophages (109). Pigmented Paget disease and pigmented epidermotropic metastatic breast carcinoma demonstrate dendritic melanocytes at the dermal–epidermal junction with variable melanin pigment in the cytoplasm of tumor cells (110). Normal numbers of melanocytes are present in depigmented extramammary Paget disease, but epidermal melanization is markedly reduced (111). In generalized hyperpigmentation from metastatic melanoma or endocrinopathy, variable epidermal pigment and melanophages are present.
Pathogenesis. Electron microscopy of hypopigmented mycosis fungoides shows degenerative changes in melanocytes. The melanocytes demonstrate dilatation of rough endoplasmic reticulum, mitochondrial swelling, incompletely melanized melanosomes, and vacuolization and disintegration of the cytoplasm (113). Giant melanosomes are seen in keratinocytes of hyperpigmented mycosis fungoides, and melanin granules are also present in the cytoplasm of tumoral and Langerhans cells (106). Fewer melanosomes are present within keratinocytes in hypopigmented sarcoidosis, and melanocytes feature variable degenerative changes (107).
Differential Diagnosis. Drug-induced hyperpigmentation.
Principles of Management. Treatment of the primary dermatosis or systemic disease.
ALBINISM
Clinical Summary. Albinism is a heritable disorder causing a generalized lack of pigmentation of the skin, hair, and eyes. Involvement of only the eyes is known as ocular albinism and is X-linked recessive. Oculocutaneous albinism (OCA) affects both the eyes and skin and is autosomal recessive. The diagnosis of OCA is primarily made through clinical findings including cutaneous hypopigmentation compared with other family members and ophthalmic findings.
There are at least 10 types of OCA based on the degree of tyrosinase activity, hair color, and associated systemic disorders. Tyrosinase is a copper-containing enzyme responsible for the biosynthesis of melanin. Mutations in the tyrosinase gene result in inactivity of the enzyme (tyrosinase-negative OCA) and cause the most severe subtype (114,115). In tyrosinase-positive OCA, synthesis of melanin pigment is reduced, and patients acquire some melanin pigment in the hair, skin, and eyes beginning in childhood. Hair color varies from yellow (yellow-mutant OCA) to red (rufous OCA) to platinum (pt OCA) to brown (brown OCA) to black (blacklocks-albinism-deafness syndrome [BADS]). Associated systemic disorders include platelet defect with mild bleeding diathesis, inflammatory bowel disease and/or pulmonary fibrosis, ceroid storage (Hermansky–Pudlak syndrome), defects in immunity (Chédiak–Higashi and Griscelli syndromes), and microphthalmia and mental retardation (Cross syndrome). Photosensitivity and cutaneous malignancies, particularly squamous cell carcinoma, are frequent in all forms of albinism.
Prenatal diagnosis of OCA can be made by analysis of fetal skin biopsy for the tyrosinase gene in fetal cells obtained by amniocentesis (116).
Histopathology. Histologic examination of skin from patients with OCA shows the presence of basal melanocytes in the epidermis and hair bulb; however, Fontana–Masson stain fails to show any melanin. In patients with the tyrosinase-positive type of albinism, the epidermal melanocytes form pigment if sections of skin are incubated with dopa.
Hermansky–Pudlak syndrome is a disorder in melanosome formation that histologically demonstrates giant melanosomes in the epidermis along with melanophages in the dermis (117–119). This syndrome is diagnosed by the absence of platelet-dense granules under electron microscopic examination. Nine human Hermansky–Pudlak genes have been identified (119,120).
Pathogenesis. OCA is a disorder of melanin synthesis within the melanosome. Electron microscopic examination in OCA shows normally structured melanocytes in the epidermis; however, there is a reduction in the melanization of melanosomes (121). For example, in tyrosinase-positive albinism, there are fewer melanized stage III and IV melanosomes, whereas in tyrosinase-negative albinism, the melanosomes contain no melanin and represent stage I and II melanosomes. Melanosome transfer to keratinocytes is not altered.
The tyrosinase gene is located on chromosome 11q 14.3 (122,123). Mutations in the protein-coding region of the gene are responsible for the tyrosinase-negative OCA. In contrast, tyrosinase-positive OCA has been mapped to the P gene on chromosome 15, and mutations of this gene are associated with a wide range of clinical manifestations (123,124).
Principles of Management. Sun avoidance, broad-spectrum sunscreen, frequent skin cancer screenings, referral to ophthalmologist (125).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


