Fig. 10.1
Schematic diagram of the sun’s radiation reaching the earth’s surface
The UV spectrum is subdivided into UVC (100–290 nm), UVB (290–320 nm), and UVA (320–400 nm) radiation. The ability of these wavelengths to penetrate the skin depends in great part on their absorption by different chromophores or photoreceptors (Fig. 10.2). UVC and UVB are readily absorbed by DNA within living cells, and thus almost all of these shorter wavelengths are absorbed as they pass through the epidermis, with little to none reaching the dermis. By contrast, UVA and visible light penetrate deeper into the dermis where these wavelengths are absorbed principally by aromatic amino acids on cell membrane proteins, and by collagen, elastin, and ground substance [1]. As will be discussed later, the development of high-energy light sources that emit 340–400 nm (UVA1) exclusively and their use as a therapeutic modality that is safer than PUVA (psoralens plus UVA) has led to its distinction from UVA2 (320–340 nm), which more closely resembles UVB. UVA1 has features intermediate between UVB and visible light. UVC and UVB are absorbed primarily by DNA, and are mutagenic. Indeed, UVC (germicidal) lamps are used to kill microbes. Of the solar radiation reaching the earth’s surface, UVB is the most carcinogenic. UVB is also the most erythemogenic and is responsible for producing sunburn and immediate tanning. By contrast, UVA is considerably less erythemogenic, but more effective in producing a longer lasting tan.
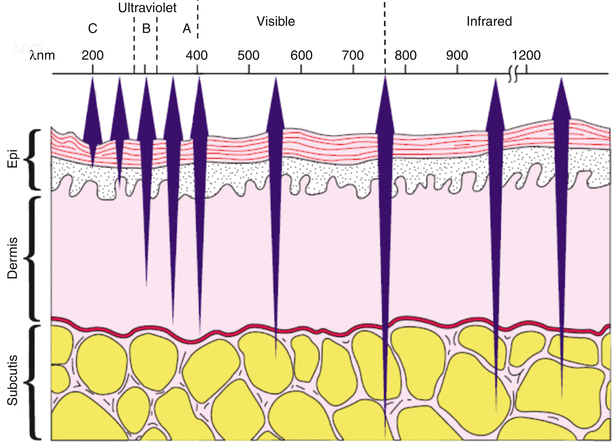
Fig. 10.2
Wavelength and depth of penetration. Within the 200- to 800-nm range, there is a direct relationship between wavelength and its depth of penetration into the skin
Photocarcinogenesis
Human epidemiologic and laboratory animal studies overwhelmingly support the concept of UV exposure as the major environmental risk factor for non-melanoma skin cancer (NMSC) [2–5]. In this respect and as cited previously, UVB is most carcinogenic [6], although large doses of UVA also have been shown experimentally to cause non-melanoma skin cancer [7]. A causative role for UV radiation in melanoma is also supported by scientific evidence, albeit at a level less substantial than for NMSC [8–10]. Both UVA and UVB may be involved in the pathogenesis of melanoma [11], but the two types of UV radiation likely act by different mechanisms. Experimentally, UVA-induced melanoma requires the presence of melanin pigment and the DNA damage is caused indirectly by reactive oxygen species, whereas UVB-induced melanoma develops in a melanin-independent manner and is due directly to UV-mediated DNA-damage [12].
UVB causes a variety of DNA mutations, most commonly cyclobutane pyrimidine dimers that have been linked to the genesis of sunburn, tanning, and skin cancer. The biologic consequences of UVB-induced mutations depend on the ability of the host to repair DNA defects and on the specific gene mutations left unrepaired. Mutations that activate oncogenes (e.g., ras) or deactivate tumor suppressor genes (e.g., p53 for squamous cell cancer; PATCHED for basal cell cancer) set the stage for carcinogenesis [13]. It is assumed that people with competent DNA repair mechanisms are able to correct UV-induced mutations. These protective mechanisms may deteriorate with age, allowing some mutations to escape repair, thereby leading to actinic keratoses, squamous cell cancers, and basal cell cancers. Xeroderma pigmentosa, a congenital disorder of absent or deficient DNA repair enzymes, provides a dramatic illustration of the foregoing concepts since afflicted individuals suffer from multiple skin cancers as early as childhood.
UV-Induced Immunosuppression
In addition to its carcinogenic effects, UV radiation promotes cancer growth by suppressing a second host defense mechanism – the ability of the immune system to kill UV-induced skin cancers. That UVB radiation leads to suppressor T-cell activation has been demonstrated not only for UVB-induced carcinogenesis but also for UVB-induced suppression of delayed-type (DTH) and contact hypersensitivity (CH) [14, 15]. The immunosuppression generated is not a generalized one, but specific for the antigen to which the host is being sensitized at the time of UV exposure [14]. Several overlapping pathways have been shown to lead to UV-induced immunosuppression (Fig. 10.3). There is overwhelming scientific evidence supporting the ability of UVB to induce immunosuppression, yet recent evidence has also suggested that UVA wavelengths from 364 to 385 nm may also be immunosuppressive [16], though the immunomodulatory effects of UVA have a complex dose–response relationship [17].
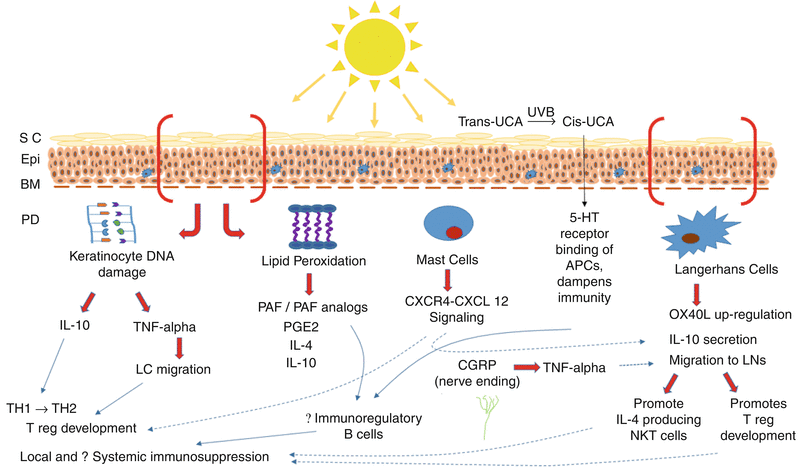
Fig. 10.3
Overlapping pathways accounting for the effects of ultraviolet radiation on the immune system. 5-HT serotonin, APC antigen presenting cell, BM basement membrane, CGRP calcitonin gene related peptide, CXCL CXC chemokine ligand, CXCR CXC chemokine receptor, Epi epidermis, IL interleukin, LC Langerhans cell, LNs lymph nodes, NKT natural killer T, OX40L ligand for CD134, also known as CD252, PAF platelet activating factor, PD papillary dermis, PGE prostaglandin E, SC stratum corneum, TH T-helper, UV ultraviolet, TNF tumor necrosis factor, T reg T regulatory cell, UCA urocanic acid
UVB light can induce keratinocytes to secrete soluble factors, of which tumor necrosis factor-α (TNF-α) [18, 19] and interleukin-10 (IL-10) [20, 21] are most critical to UVB immunosuppression; TNF-α regulates Langerhans cells’ emigration out of the epidermis into draining lymph nodes, and IL-10 shifts T-cell responses from T-helper-1 (Th1) to Th2 phenotype [20]. UV light can also trigger lipid peroxidation on cell membranes leading to secretion of platelet aggregation factor (PAF), which in turn stimulates prostaglandin E2 (PGE2) production, which in turn prompts IL-4 secretion, and also IL-10 production [21, 22]. UV-induced oxidation reactions can also produce PAF analogs such as 1-alkyl-2-(butanoyl and butenoyl)-sn-glycero-3-phosphocholine that can stimulate the PAF receptor [23]. Trans-urocanic acid, a by-product of histidine metabolism, accumulates in the stratum corneum; UV non-enzymatically transforms trans-urocanic acid into the cis isomer, which contributes to immunosuppression via effects on antigen-presenting cells (APCs) [24, 25]. Immunosuppressive effects of cis-urocanic acid were shown to be mediated through binding to the serotonin (5-HT) receptor [26]. Finally, UVB radiation can trigger release of calcitonin gene related peptide (CGRP) from cutaneous nerve endings, leading to mast cell release of TNFα, which dampens CH responses [27].
UV can directly alter the function of epidermal Langerhans cells (LC) and other APCs. Depending on the UV dose and the manner in which it is administered, UV may induce these APCs to undergo apoptosis, shift their function from stimulators of Th1 (cellular) to Th2 (humoral) responses, or cause them to activate “suppressor” T cells [28–30]. Previously, as LC were regarded as the most important APCs in the skin, it was thought that such antigen tolerance was due to epidermal LC depletion by UV light. [31] Now dermal dendritic cells (FITC+, CD11c+, Langerin−) are thought to be critical for antigen presentation and immune stimulation (even in the setting of UV light) [32, 33]. By contrast, LC play a significant role in UV-mediated immunosuppression. UVB radiation leads to LC maturation, up-regulation of surface marker OX40L, and production of IL-10, which upon migration to lymph nodes, supports the development of regulatory T cells [34]. Regulatory T cells express CD4, CD25, and CTLA-4 markers, secrete copious amounts of IL-10, bind dectin-2, and dampen immune responses [35, 36]. LC migrating to the lymph nodes may also mediate immunosuppression by activating a population of natural killer T (NKT) cells to secrete IL-4 [37]. Interestingly, topically applied low-dose green tea extract (3 %) has recently been shown to reduce UV-mediated depletion of LC [38].
Additionally, mast cells and complement activation via the C3 pathway have been shown to participate in UVB-induced immunosuppression [39–42], and evidence continues to mount in support of an integral role for mast cells in UV-induced immunosuppression [43]. Dermal mast cells have been shown to critically mediate UV-induced immunosuppression through the CXCR4-CXCL12 signaling pathway. In fact, blockade of mast cell trafficking with the CXCR4 antagonist AMD3100 prevented UV-induced immunosuppression and reduced the number of cutaneous squamous cell carcinomas in mice, presumably by reducing mast cell migration into local lymph nodes and the tumors themselves [44, 45]. Interestingly, studies have shown an increased mast cell density in buttock skin of patients with basal cell carcinoma and melanoma compared to controls [46, 47].
UV light also leads to the development of a suppressive population of B cells that express high levels of major histocompatibility complex II (MHC II) and B220 but low levels of co-stimulatory molecules. When dendritic cells and B cells isolated from UV-radiated mice were conjugated to antigen ex vivo and injected into naïve hosts, this subset of B cells suppressed dendritic cell activation of immunity (which occurred in the absence of these B cells) [48]. Both platelet activating factor (PAF) and serotonin were shown to promote development of these so-called “immunoregulatory B cells” (FITC+, IL-10-secreting, CD19+, B220+) capable of inducing tolerance to antigens following UV exposure [49].
In addition to attenuation of CH and skin cancer immunosurveillance, UV light has been shown to serve as an immunomodulator for systemic autoimmune diseases, including multiple sclerosis, type I diabetes, and rheumatoid arthritis [50]. Much of the evidence is epidemiologic: the latitude gradient in these diseases suggests an inverse relationship between disease incidence and UV exposure. However, in a murine model of multiple sclerosis (experimental autoimmune encephalitis or EAE), UVB radiation was shown to induce tolerogenic dendritic cells that circulated systemically and promoted development of regulatory T cells in several organs, including the CNS [51].
UVB Radiation and Vitamin D
UVB causes the non-enzymatic conversion of 7-dehydrocholesterol to cholecalciferol (vitamin D3) in skin. Although vitamin D is well-known for its effects on mineral metabolism and bone health, it has also become apparent that cholecalciferol modulates immune responses in skin and, perhaps, systemically. Vitamin D can modulate UVB-mediated damage to keratinocytes, although the effect is lost at higher doses of UVB irradiation, which may explain increased skin cancer risk associated with chronic, high dose UV exposure [52]. Several laboratory studies have suggested a protective role of vitamin D against basal cell and squamous cell carcinoma as well as melanoma, although further human studies need to be performed. Toll-like receptors (TLRs) activated in response to intracellular bacteria can upregulate vitamin D receptors on macrophages, leading to the induction of canthelicidin and intracellular killing of Mycobacterium tuberculosis [53]. In addition, sera from African Americans, a population known to be more susceptible to tuberculosis, were found to have lower levels of 25-hydroxyvitamin D compared to controls. Vitamin D may also stimulates phagocytosis by monocytes and macrophages [54].
While these examples support an immunostimulatory role for vitamin D, there is also data suggesting that vitamin D may be immunosuppressive. Cholecalciferol may inhibit the production of Th1 type cytokines, suppress activation of Th1 cells, and enhance the function of regulatory T cells [50]. These actions may play be protective or preventative of autoimmunity. Life-long vitamin D supplementation in NOD mice prevented development of diabetes mellitus whereas induced vitamin D deficiency from birth led to early development of type I diabetes mellitus [55]. A promising role for UVB radiation in multiple sclerosis is related to elevated levels of vitamin D in patients treated with UV light. [51] The exact role of vitamin D in skin and systemic immunity remains to be fully elucidated. UVB as the primary external stimulant of vitamin D production has the potential to mediate vitamin D- dependent immunity.
Immunologically-Mediated (Idiopathic) Photodermatoses
The immunologically mediated photodermatoses (IMPs), also known as idiopathic photosensitive skin disorders, are a heterogeneous group of conditions for which exact etiopathologic mechanisms remain elusive though each manifests immunologic underpinnings (Table 10.1). Our focus will be on polymorphic light eruption, actinic prurigo, solar urticaria, hydroa vacciniforme, and chronic actinic dermatitis.
Table 10.1
Photosensitivity disorders
Immunologically-mediated (idiopathic) photodermatoses | Polymorphic light eruption (including juvenile spring eruption) |
Actinic prurigo | |
Solar urticaria | |
Hydroa vacciniforme | |
Chronic actinic dermatitis | |
Defective DNA repair | Xeroderma pigmentosa |
Trichothiodystrophy | |
Cockayne syndrome | |
Rothmund-Thompson syndrome | |
Autoimmune | Lupus erythematosus |
Dermatomyositis | |
Drug-induced | Phototoxic reaction |
Photoallergic dermatitis | |
Pseudoporphryia | |
Porphyrias | Porphyria cutanea tarda |
Erythropoietic porphyria |
Polymorphic (Polymorphous) Light Eruption
Polymorphic light eruption (PMLE) is the most common photosensitivity disorder, with estimates of prevalence as high as 20 % of the general population [56, 57]. PMLE has a predilection for women, an age of onset in the teenage and young adult years, and a tendency to affect people living in temperate climates. Recurrent outbreaks typically occur in spring, with clinical onset occurring several hours following UV exposure. The eruption develops over sun-exposed skin, is usually pruritic, and may take several morphologic patterns (i.e., polymorphic): small to large papules, vesicles, plaques, and even erythema multiforme-like eruptions [56]. Despite the wide variety of morphologic patterns, individual patients tend to present with the same morphologic feature in a recurrent manner [58]. Outbreaks typically last 1–2 weeks, and the eruption frequently improves in the summer (termed hardening). Photoprovocation studies have implicated different wavelengths spanning UVB, UVA, and even visible light ranges [59, 60]. Juvenile spring eruption (JSE) is considered a localized variant of PMLE that tends to occur on the ears of young boys. Outbreaks most commonly occur during the spring after exposure to bright sunlight in cold weather, with body sites lacking hair (e.g., protuberant ears) as favored locations [61].
Pathogenesis is believed to be a delayed cellular hypersensitivity reaction to an endogenous cutaneous antigen possibly generated by the action of UV radiation [62, 63]. Although the identity of the chromophore/antigen remains unknown, studies have shown patients to be relatively incapable of manifesting UVB-induced suppression of immune responses like induction (but not elicitation) of contact hypersensitivity [64], a defect ascribed to inability of UVB to induce emigration of Langerhans cells from the epidermis to draining lymph nodes [65, 66]. Lesions of PMLE have demonstrated an altered immunoregulatory network, with reduced epidermal or dermal expression of TGF-beta-1, IL-10 and RANKL, and a relatively low number of Tregs [67]. This reduced ability of UVB to suppress immune responses leads to an enhanced immune response to cutaneous neoantigens generated from UV exposure. Repeated UV exposure in these patients eventually suppresses immunity, explaining the natural improvement (hardening) during summer months as well as the therapeutic role of prophylactic NB-UVB or PUVA therapy [58].
Most cases of polymorphic light eruption are mild and can be managed by photoprotection plus topical corticosteroids for symptomatic relief. Occasionally in young, otherwise healthy patients, a short course of systemic corticosteroids (0.6 mg–1 mg/kg per day for 1 week) can help control symptoms. For patients with severe, recurrent disease, NB-UVB or PUVA therapy in late winter can prevent seasonal flares during spring [58].
Actinic Prurigo
Actinic prurigo is an immune-mediated photodermatosis most commonly seen in the indigenous Indian and mestizo populations of Central and South America, especially individuals living at high altitudes, although Caucasian and Asian populations are also affected [58]. The disorder is thought to be caused by transformation of an epidermal antigen upon exposure to UV light, leading to an abnormal immune response [68]. Although both UVA and UVB have been implicated in actinic prurigo, the eruption is most often linked to UVA radiation [58, 68]. Typically, actinic prurigo presents in children younger than 10 years (often 4–5 years) with intensely itchy papules, plaques and nodules as well as excoriations and scarring on sun exposed areas. The face, neck, extensor forearms, dorsal hands, and upper chest are usually affected, but involvement of the back and buttocks may occur. Conjunctivitis and cheilitis (lower > upper lip) are characteristically present. Pruritus tends to last throughout the entire year, with exacerbations in the spring and summer.
While actinic prurigo may share with polymorphic light eruption a similar immunologic mechanism, lesion morphology, and delayed-onset relative to sun exposure, there are several clinical differences between the two disorders. As outlined above, actinic prurigo is more likely to affect both exposed and covered sites, can occur in the winter, tends to persist beyond 4 weeks, often exhibits lip or conjunctival involvement, and frequently presents with excoriation and scarring [69]. Furthermore, actinic prurigo has been linked to human leukocyte antigen (HLA) subtypes. Originally HLA subtypes A24 and Cw4 were linked to actinic prurigo in patients of Cree ancestry [70], but subsequent studies in both European and Central American populations demonstrated strongest association with the DR4 allele, with the specific allele HLA-DR4/DRB1*0407 being most common, found in 60–70 % of affected individuals [58, 68, 69, 71].
Unlike polymorphic light eruption, management of actinic prurigo tends to be more challenging. Photoprotection is the most important measure, including use of protective clothing, sunscreens, lip balms, and sunglasses. Multiple therapies may reduce pruritus, including topical corticosteroids, emollients, and oral antihistamines. Occasionally NB-UVB and PUVA are also used, although the level of evidence for their efficacy is lower than for polymorphic light eruption [58]. In actinic prurigo, the most consistently effective form of therapy is thalidomide, although its use is limited by adverse effects including teratogenicity, peripheral neuropathy, and venous thromboembolism [58, 68, 72].
Solar Urticaria
Solar urticaria is a rare form of physical urticarial provoked by exposure to UV light. Most studies support an action spectrum between 300 and 500 nm, although the exact action spectrum tends to vary between studies and individuals. Rare cases due to infrared light have also been reported [73, 74]. Solar urticaria affects about 0.8 % of patients with urticarial eruptions, and up to 7 % of patients with photodermatoses. The disorder has a variable age of onset, although most patients present in young adulthood. Women tend to be affected more commonly than men. There does not seem to be a racial or ethnic predilection [73].
Unlike polymorphic light eruption and actinic prurigo, solar urticaria typically develops within minutes of exposure to sunshine and manifests as itching, burning, erythema, and wheals [75]. The eruption tends to affect “classic” photo-distributed areas such as the V-shaped area of the upper chest and the arms, but urticarial lesions may spare areas regularly exposed to sunlight like the face and hands. Systemic symptoms including headache, nausea, wheezing, dizziness and rarely anaphylactic shock [73].
Solar urticaria is an immediate type of photoallergic reaction caused by immunoglobulin E (IgE) autoantibodies. There are two subtypes of solar urticaria based on our most current understanding of these autoantibodies. In Type 1 solar urticaria, affected individuals have an abnormal chromophore within the skin to which IgE antibodies develop, leading to urticarial lesions. In these cases, passive transfer of serum to a normal individual will variably produce lesions of solar urticaria upon light exposure if the abnormal chromophore is also contained in the serum. By contrast, in Type 2 solar urticaria, affected individuals have IgE antibodies to a normal chromophore in the skin. In these cases, passive transfer of serum to a normal individual will always lead to urticaria upon subsequent light exposure [73]. While this distinction is useful for understanding underlying mechanisms of solar urticaria, passive transfer experiments are not routinely performed for ethical reasons, so the distinction between these subtypes is generally not made in clinical practice.
Treatment of solar urticaria is aimed at strict photoprotection. As in other types of urticaria, symptom relief can be achieved with topical corticosteroids or oral antihistamines. Although phototherapy should be approached with caution due to potential flaring of the disease, both NB-UVB (only to areas of skin that are regularly sun exposed, to produce “hardening”) and PUVA have been used effectively in select cases. There is limited evidence supporting the use of beta-carotene, antimalarial medications, plasmapheresis, systemic immunosuppressives, and intravenous immunoglobulin [58, 62].
Hydroa Vacciniforme
Hydroa vacciniforme is a rare photosensitivity disorder found almost exclusively in children, with a mean age of onset around 8 years. Boys are affected more often than girls [58, 76]. Most patients are sensitive to UVA radiation, which causes recurrent crops of 2- to 3-mm erythematous macules that evolve into blisters within a few hours to days. The lesions then become umbilicated with crusting within a few days, often mimicking impetigo or impetiginized herpes simplex virus infection. These lesions heal with pitted varioliform scarring. The face and hands are the most commonly affected sites.
Epstein-Barr Virus (EBV) infection has been linked to hydroa vacciniforme. One case report of EBV-associated hydroa vacciniforme suggested that plasmacytoid monocytes (CD68+ and CD123+) may play a crucial role in the pathogenesis based on immunohistochemistry from the patient’s biopsy demonstrating a predominance of this cell type [77]. More recently, a case series evaluating biopsies and peripheral blood of patients with hydroa vacciniforme found an increase in EBV-infected γ/δ T cells in affected patients which was not present in controls with hypersensitivity reactions to mosquito bites [78]. However, the exact pathophysiology to explain the relationship between the viral infection and UVA light is yet to be elucidated.
No treatments for hydroa vacciniforme are consistently successful, so photoprotection remains a critical aspect of management. Occasionally anti-malarials, beta-carotene, PUVA, systemic immunosuppressives, and thalidomide have been used successfully [62]. In severe cases, systemic corticosteroids can be used [58].
Chronic Actinic Dermatitis
Chronic actinic dermatitis is an umbrella term for several forms of chronic photosensitivity disorders including photosensitive eczema, persistent light reaction, chronic photosensitive dermatitis, and actinic reticuloid. It often begins insidiously as nonspecific eczematous lesions in sun-exposed areas that become more persistent, in the absence of known topical or systemic photosensitizers [62]. Eczematous lesions are by far the most common, though papular lesions and pseudolymphomatous “reticuloid” lesions may also occur [79]. It is more common in men and tends to occur in later adult years, although it may occur earlier in patients with a strong history of atopic dermatitis [56, 80]. UV-B, UV-A, and visible light, either alone or in combination with each other, have been implicated in chronic actinic dermatitis [81].
Although the exact etiology is uncertain, the cause appears to be a form of delayed type hypersensitivity to a normal skin constituent that becomes altered by UV light and becomes antigenic in the absence of “normal” UVB-induced immunosuppression [80–82]. In many cases, there is a strong association with allergic contact dermatitis, though it remains unclear whether ACD is directly involved in the pathogenesis of chronic actinic dermatitis or whether allergy develops secondary to abnormal barrier function in the setting of a susceptible cutaneous microenvironment. Sesquiterpene lactone has a well-known association with chronic actinic dermatitis and remains an important allergen even in recent studies. Other studies have shown a significant increase in patch test positivity to non-fragrance consumer allergens in affected individuals, particularly para-phenylenediamine [83]. Up to 75 % of patients react on patch testing or photopatch testing to allergens suspected to be involved in chronic actinic dermatitis; it is unclear whether the remaining patients have underlying allergy to an untested hapten or develop chronic actinic dermatitis in the absence of allergic contact dermatitis [80, 83].
With appropriate treatment, including avoidance of sunlight, regular use of sunscreen and skin-protective clothing, and avoidance of known allergen(s), chronic actinic dermatitis tends to improve or resolve over the course of many years, with approximately 55 % of patients experiencing improvement and 35 % experiencing resolution over 15 years in one study [84]. Aside from these basic measures, many other therapeutic options may be tried, including emollients, topical corticosteroids, topical tacrolimus, systemic corticosteroids, azathioprine, cyclosporine, methotrexate, hydroxyurea, retinoids, antimalarials, and phototherapy. Azathioprine tends to be quite effective for severe forms of this disorder, and systemic corticosteroids may be beneficial in acute exacerbations. Phototherapy tends to be poorly tolerated, but has been useful in some patients [79].
Phototesting and Photopatch Testing
Both phototesting and photopatch testing may be diagnostically useful in the evaluation of immunologically-mediated photodermatoses. Sensitivity to UV is usually assessed by measuring the minimal erythema dose (MED), which is the amount of UV radiation that will produce minimal erythema. MED can be tested with either UVB or UVA wavelengths but tends to be more reliable for evaluating photosensitivity to UVB. In PMLE, most patients have normal MEDs to UVB and UVA, although some patients have decreased MEDs to UVB and/or UVA. However, photoprovocation testing (repeated exposures to sub-erythemogenic doses of UV radiation for 3–4 days) often reproduces the eruption [58]. Although not necessary for diagnosis, phototesting may be useful in the evaluation of other immunologically-mediated photodermatoses (Table 10.2). Photopatch testing is a procedure in which a typical patch test is performed with addition of UV exposure (usually UVA) upon removal of patches. In addition to evaluation of photo-allergic contact dermatitis, photopatch testing can be useful in the diagnosis of chronic actinic dermatitis [81].
Table 10.2
Evaluation of photosensitivity disorders
Disorder | Phototesting | Photopatch testing | ||
|---|---|---|---|---|
Useful? | Result? | Useful? | Result? | |
Polymorphic light eruption | Yes | Normal (usually) or decreased MEDs to UVB/UVA | No | N/A |
Actinic prurigo | Yes | Low or normal MEDs to UVA > UVB | No | N/A |
Solar urticaria | Yesa | Broad spectrum sensitivity (hives) to visible and UV light | No | N/A |
Hydroa vacciniforme | Yes | Vesiculation/scarring following UVA exposure | No | N/A |
Chronic actinic dermatitis | Yes | Delayed dermatitis or pseudolymphomatous response to UVB, UVA and visible light | Yes | Delayed dermatitis, often to sunscreen allergens, topical medicines and Compositae |
Phototherapy
Along with Mohs micrographic surgery for the treatment of skin cancer and patch testing for the diagnosis of allergic contact dermatitis, phototherapy is among the most quintessential of dermatologic procedures (rarely performed by other medical specialists). Phototherapy refers to the use of artificial UV light to treat skin (and potentially systemic) disorders, a domain that for several decades was based largely on empirical evidence but over time has developed a stronger foundation in the scientific literature.
The major mechanisms of phototherapy in most inflammatory skin disorders, such as psoriasis and atopic dermatitis, are: (1) diminishing effector T cell responses, (2) promoting the development of regulatory immune cells, and (3) restoring normal barrier function, especially in atopic dermatitis [85]. Increasing knowledge and insight derived from photoimmunology have provided cellular and molecular mechanisms that account for phototherapy’s beneficial effects (Table 10.3). Both UVB and UVA1 can induce apoptosis, and the threshold for causing programmed cell death is much lower for lymphocytes than for other cells. Apoptosis by UVB may be achieved via mutations or activation of death receptors (e.g., Fas/Fas ligand, TNF receptor, TRAIL [TNF-Related Apoptosis Inducing Ligand] receptor) [86]. By contrast, apoptosis by UVA may be mediated through reactive oxygen species, differential killing of Th1 over Th2 cells, or lysis of mast cells [86]. As cited previously, UVB light can induce secretion of cytokines including IL-1, IL-4, IL-6, IL-8, IL-10, and TNF-α, with the last two deemed responsible for immunosuppression [18–22]. UV light also leads to down-regulation of major histocompatibility complex (MHC) and co-stimulatory molecules on antigen presenting cells (APCs) in the skin, making these cells functionally impaired in their ability to stimulate T cell effector function [85]. As cited previously, ultraviolet B also generates regulatory immune cells, the most well-characterized being regulatory T cells which are critical to phototherapy-mediated immunosuppression [34–36]. Regulatory T cells are FoxP3+ and CD4+, and they secrete IL-10 and TGF-beta, which dampen effector T cell and APC function. By contrast, UVA (but not UVB) can inhibit collagen synthesis and metalloproteinase activity, leading to changes in extracellular matrix which makes UVA therapy useful in sclerotic skin diseases [86].
Table 10.3




Mechanisms of phototherapy
Related posts:





Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
