Fig. 4.1
The clinical stages of mycosis fungoides
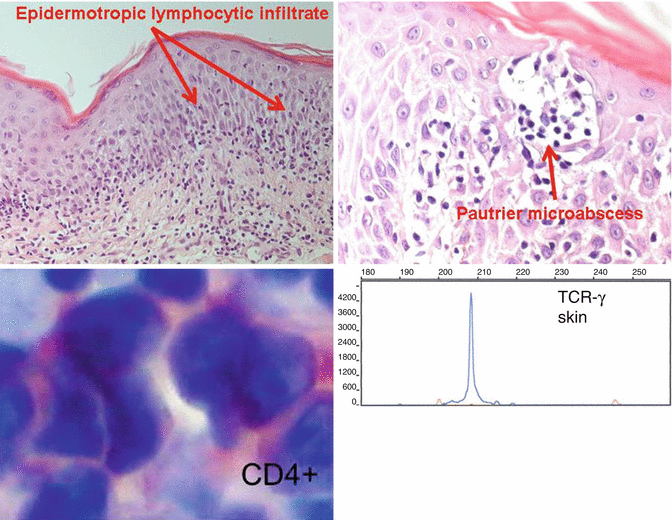
Fig. 4.2
Histopathological and molecular diagnosis of mycosis fungoides
Table 4.1
WHO classification of cutaneous lymphomas
Cutaneous T-cell and NK-cell lymphomas | Cutaneous B-cell lymphomas |
|---|---|
Mycosis fungoides (MF) | Primary cutaneous follicular B-cell lymphoma (PCFCL) |
Mycosis fungoides variants and subtypes | Primary cutaneous marginal zone B-cell lymphoma (PCMZL) |
Folliculotropic MF | Primary cutaneous diffuse large cell B-cell lymphoma – leg type (PCBLT) |
Pagetoid reticulosis | Primary cutaneous diffuse large cell B-cell lymphoma, others |
Granulomatous slack skin | |
Sézary syndrome (SS) | Primary cutaneous intravascular large cell B-cell lymphoma |
Adult T-cell leukemia/lymphoma | |
Primary cutaneous CD30+ lymphoproliferative diseases | Hematological precursor neoplasms CD4+, CD56+ hematodermic neoplasm (plasmacytoid dendritic cell neoplasm) |
Primary cutaneous anaplastic large cell lymphoma (PCALCL) | |
Lymphomatoid papulosis (LyP) | |
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) | |
Extranodal NK/T-cell lymphoma, nasal type | |
Primary cutaneous γ/δ T-cell lymphoma | |
Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (provisional) | |
Primary cutaneous small- to medium-sized pleomorphic T-cell lymphoma (provisional) | |
Peripheral T-cell lymphoma, not otherwise specified |
CTCL challenges both basic and clinical research for three reasons: first, its pathogenesis is widely unknown, although evidence on altered signaling or metabolic pathways as well as changes in the microenvironment increases. Second, quick and precise diagnosis is complicated by the lack of unique diagnostic markers defining CTCL and demarcating it from other skin diseases. Third, the disease is not curable by now. Existing CTCL treatment only prolongs or delays the disease progression and shows high relapse rates. Hence, there is an urgent need to further investigate the mechanism of CTCL development in order to develop new therapeutic approaches.
4.2 New Insights into CTCL Pathogenesis
The development of individualized therapeutic approaches requires knowledge on the pathogenesis of a disease. Information is needed on possible alterations in signaling or metabolic pathways, in microenvironment or protein activities in a specific patient. Identifying any cellular or molecular changes that might explain the development of the disease is essential, as every difference between a malignant and a healthy cell represents a possible therapeutic target. In recent years, a wide variety of altered molecular structures or pathways have been identified that might be therapeutically targeted. Each of these altered target structures affects only a more or less small percentage of CTCL patients, which underlines the need for individualized approaches in identifying target structures for cancer therapy. The development of such therapies fills a gap in the therapeutic spectrum of CTCL that at the moment consists mainly of general therapeutic concepts.
4.2.1 Apoptosis Resistance
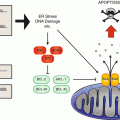
The malignant T-cell population in CTCL is characterized by a phenotype of resting, CD3+CD4+CD7−CD45RO+-bearing Th2 memory cells. The lifespan of a peripheral T cell is normally limited to several days under physiologic conditions. Upon activation, a cell death program, the activation-induced cell death (AICD), is enabled and even decreases this lifespan. CTCL cells have been found to return to a resting state decreasing their turnover and rendering them insensitive towards cell death stimuli. They evade AICD that is triggered by the death receptor CD95 by their lack of phospholipase-γ1 [24]. This enzyme is needed to process the TCR stimulation signal intracellularly that leads to formation of CD95 ligand. This ligand activates CD95 in an autocrine way leading to induction of AICD (Fig. 4.3). Due to this mechanism and their resistance towards intrinsic and extrinsic cell death stimuli, the accumulation of CTCL cells in the skin and subsequently in the lymph nodes rather derives from an acquired resistance towards cell death than from an increased proliferation rate. This cell death resistance is a very typical characteristic of CTCL cells that originates from alterations in several signaling pathways. Restoring sensitivity towards cell death contains a powerful therapeutic potential in fighting CTCL.
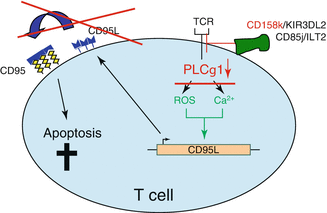
Fig. 4.3
Modified TCR signaling is responsible for AICD resistance in CTCL
The nuclear factor κB (NFκB) represents one important trigger structure that can turn a cell resistant to cell death stimuli. Different alterations in this complex pathway have been described in CTCL cell lines and primary patient cells. These alterations lead to a constitutive activation in the NFκB pathway in the nucleus which impairs cell death signaling. The exact reason for this activation is unknown, but enhanced levels of single NFκB subunits like p50 as well as increased DNA binding of NFκB components found in CTCL cells explain it at least in part. This insight into CTCL pathogenesis has led to great ambition in medically reverting NFκB activation and thus inducing apoptosis in the CTCL cells [21, 36].
NFκB activity can be targeted directly and indirectly via either inhibiting NFκB components themselves or other molecules crosstalking with the NFκB pathway. NFκB activation is sensitively regulated by phosphorylation and dephosphorylation of single components. The kinases in this interplay are well characterized. Recently, the phosphatases involved in antagonizing the kinase activity have come into focus more closely. The phosphatase PP4R1 connects the inhibitory phosphatase PP4c with the IKK complex within the NFκB signal, so that NFκB signaling is inactivated by PP4c. PP4R1 is hardly expressed in malignant T cells of many CTCL patients, so that PP4c cannot bind to the IKK complex and thus block its signaling (Fig. 4.4). This results in a constitutive activation of the NFκB pathway [6]. This phosphatase activity might represent an interesting therapeutic target for an individualized approach, but by now no substance exists that would interfere with the mentioned mechanism.
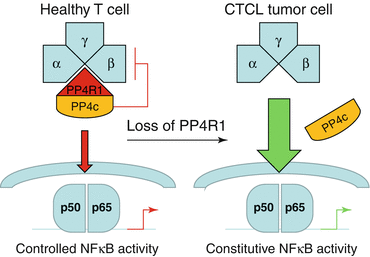
Fig. 4.4
Scheme of NFkB phosphatase signaling in healthy and CTCL T cells
Another pathway that is related to the NFκB pathway and can also, independently of NFκB, lead to enhanced survival signals in T cells is the MAPK pathway. This signaling cascade has also been recently found relevant for CTCL pathogenesis and represents another structure for individualized CTCL patient care for several reasons. Both in CTCL cell lines and in isolated T cells of about 5 % of CTCL patients single nucleotide mutations in the n-ras (Q61K) and k-ras (G13D) genes were detected. These mutations lead to an excessive activation of the gene products N-Ras and K-Ras within the MAPK pathway and thus to decreased cellular susceptibility towards cell death signals [22]. As the MAPK and the NFκB pathway communicate, enhanced MAPK activity also results in increased constitutive NFκB activity.
4.2.2 Altered T-Cell Functions
Besides cell death resistance, other T-cell functions have been found impaired in CTCL cells on a molecular level recently. In parallel to other malignancies like melanoma, the expression of the cell surface protein programmed death-1 (PD-1) is pathologically increased in some types of CTCL [9, 20]. Similar to the surface protein CTLA-4 PD-1 inhibits effector T-cell function and thus possibly leads to suppression of an antitumor immune response. However, the exact significance of PD-1 overexpression is not clarified in the context of CTCL pathogenesis. Nevertheless, PD-1 can already be targeted medically; respective studies are in the clinical phase in melanoma patients. So this overexpression in CTCL contributes to the spectrum of targets for individualized approaches in CTCL therapy.
In addition, a wide variety of molecules influencing cellular transcription patterns have been found altered in CTCL. For example, the transcription factor E2A is genetically deleted in about 70 % of patients with Sézary syndrome. This loss results in increased proliferation rates and cell cycle progression via derepression of the proto-oncogene MYC and the cell cycle regulator CDK6 [38]. The transcription factor E2A also communicates in a synergistic with the Ras pathway that also enhances CTCL cell survival. Transcriptional activity is further pathologically influenced by signal transducer of transcription (STAT) protein family members in CTCL. STAT3 shows constitutional activation in Sézary syndrome, at least in part due to autocrine IL-21 stimulation [42]. STATs trigger the expression of certain miRNAs like miRNA155 and miRNA21 in CTCL [26, 43] which themselves induce resistance towards cell death stimuli.
A unique target for T-cell-directed therapy in CTCL is found in the T-cell receptor (TCR). The monoclonal, malignant T-cell population bears a specific TCR composition that differs from the TCR repertoire that is formed by the nonmalignant cell population. So far, one primary limitation in this approach lies within the detection of the monoclonal TCR. In recent years, the detection methodology has largely improved including highly sensitive PCR and high-throughput deep sequencing techniques [44].
4.2.3 CTCL Tumor Microenvironment
Basic cancer research has mainly focused on the tumor cell itself. Since the first description of the hallmarks of cancer by Hanahan and Weinberg in 2000, it became more important to investigate the microenvironment of each tumor [17]. Today, it is known that tumor cells interact with their microenvironment in order to avoid immune destruction and to induce tumor-promoting inflammation [18]. The interaction of CTCL tumor cells with inflammatory cells in the skin is also important for the understanding of CTCL pathogenesis. In many CTCL skin lesions, the minority of skin-infiltrating cells represent CTCL tumor cells. The majority of lesional cells are reactive immune cells – mainly lymphocytes. Only in the rare case of progressive advanced disease the tumor cells become more prominent than the reactive cells. Therefore, CTCL basic research has so far mainly focused on the CTCL tumor cell microenvironment.
Tumor-infiltrating inflammatory cells are known to stimulate tumor cell growth, to induce angiogenesis in tumors, and to promote tumor invasion as evidenced by a dense inflammatory infiltrate at the marginal zones of a tumor. Immune progenitor cells (CD11b+, Gr1+) are able to suppress cytotoxic T and NK cells directed against the tumor. Rabenhorst et al. investigated the role of mast cells in the pathogenesis of CTCL [32]. They observed a better outcome in CTCL patients with less mast cells in the CTCL lesion (<100 mast cells/mm2) than those with a denser cutaneous mast cell infiltrate (>100 mast cells/mm2). Their in vitro experiments identified a stimulation of CTCL tumor cell growth by incubation with a mast cell supernatant. They concluded a growth promotion of CTCL tumor cells by mast cells.
In 1990, Reinhold et al. isolated suppressive lymphocytes from MF skin lesions in contrast to non-suppressive MF tumor cells [33]. Five years later, these suppressive cells were termed regulatory T cells (Treg) [35]. Treg are CD4+ lymphocytes with suppressive properties in order to terminate immune responses and to avoid autoimmunity. They are characterized by expression of the transcription factor FOXP3. Elimination of these important immune regulatory cells leads to severe autoimmune conditions in mice and humans. Immunohistochemical studies of CTCL skin samples revealed a significant decrease of FOXP3+ cells in biopsies from Sézary syndrome in contrast to MF lesions [25]. Other studies correlated low numbers of FOXP3+ cells with advanced disease in MF or CD30+ cutaneous lymphoma patients (summarized in [28]). The physiological target of suppression by Treg is the CD4+ lymphocyte. The abovementioned findings in CTCL lead to the hypothesis that Treg are capable of suppressing CTCL tumor cells in the early stages. The property is lost in advanced disease or aggressive forms of CTCL (e.g., Sézary syndrome) (Fig. 4.5).
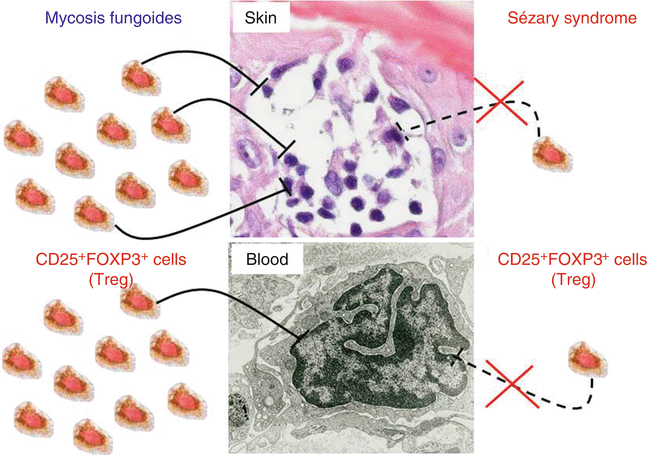
Fig. 4.5
The number of Treg correlates with an indolent course in CTCL
4.3 Personalized Aspects in Established CTCL Therapies
4.3.1 Standard of Care/Treatment Guidelines
Treatment of CTCL patients is entity specific due to the very different clinical courses of the different types of CTCL (see Fig. 4.1 and Table 4.1). And treatment should be stage-adapted to avoid too aggressive therapies. Most patients run an indolent course and require treatment for years up to decades. Therefore, the therapeutic effect needs to be balanced against possible side effects. The treatment guidelines of the German dermato-oncology group recommend skin-directed therapies for indolent forms and early stages [37]. Skin-directed therapies include topical corticosteroids, UV-based treatments like PUVA or UVB311 nm and radiotherapy. In more advanced disease or nonresponders, systemic treatments are added to the skin-directed treatment. First-line systemic treatments are interferon-α and the rexinoid bexarotene. In erythrodermic patients, extracorporeal photopheresis is given additionally or as monotherapy. For second-line treatments of CTCL, either cytotoxic monotherapies like low-dose methotrexate, gemcitabine, or liposomal doxorubicin or antibodies like denileukin diftitox are applied. Other options include the use of HDAC inhibitors like vorinostat and romidepsin or total skin electron beam irradiation. In the majority of CTCL patients, the disease can be well controlled with the mentioned standard treatments. However, even within one type of CTCL, individual patients might behave very differently requiring personalized treatment approaches.
4.3.2 Individualized Management of Bexarotene Therapy
Although being a standard therapy bexarotene treatment requires an elaborate personalized therapeutic regimen. Its characteristic side effects are often misunderstood and may lead physicians to wrong interpretations and decisions. Mainly these side effects include central hypothyroidism and hyperlipidemia, which are observed in almost 100 % of patients [1]. The occurrence of these side effects can be explained by the molecular mechanism of the bexarotene effect. This compound mainly affects nuclear retinoid X receptors (RXR) and thus influences apoptosis. RXR are in combination with retinoic acid receptors (RAR) involved in the regulation of thyroid function and lipid metabolism. So intriguingly, hypothyroidism and hyperlipidemia rather reflect bexarotene being active and effective in a patient than displaying a reason for discontinuation of treatment. Both bexarotene effectiveness in suppression of CTCL symptoms and the occurrence of these side effects show great interindividual differences, but so far there are no known correlations with any individual parameters that would allow any predictions about effectiveness and severity of side effects in a certain patient. Therefore, the side effect management has to be tuned individually in accordance with their characteristic and severity. All patients need an accompanying medication with thyroxin, whereas the dosage may vary between 50 and 200 μg/day depending on the blood values of thyroxin and triiodothyronine. The dramatically suppressed TSH that necessarily occurs during bexarotene therapy reflects the central character of the hypothyroidism and may in combination with the normal values of thyroxin and triiodothyronine under substitution not be mistaken for a constellation of peripheral hyperthyroidism. The individualized management of hyperlipidemia is more sophisticated. First-line therapy would include fibrates in therapeutic doses. In case of insufficiency of this therapy, it can be combined with esterified fish oils that are FDA approved under the brand name Lovaza® or Omacor® to lower very high triglyceride blood levels. In case of myopathies or signs of rhabdomyolysis under fibrate therapy, it can be replaced by atorvastatin 10 mg/day that influences both triglyceride and cholesterol levels with a milder side effect spectrum [1, 14, 39]. Obviously dose adjustments have to be considered in accordance with efficacy and risk of the treatment. In our experience, not many patients tolerate the maximum dose aimed for, which is not necessarily a problem. Most patients who respond at all to bexarotene show already good response rates at moderate doses, so that tapering down the dose to the lowest level of response is obviously the most reasonable method to manage side effects. It has to be emphasized that bexarotene needs up to 6 months to show a clinical therapeutic effect.
4.3.3 Individualized Management of Interferon Therapy
The handling of interferon therapy with respect to side effect management or concurrent medication is less difficult and requires less experience and consideration of mechanistic interaction. In terms of concurrent medication, the most common side effects that have to be individually dealt with are flulike inflammatory responses and depressive disorders.
The first one can in almost all cases easily be treated by nonsteroidal anti-inflammatory drugs (NSAID) like paracetamol 500 mg 1 h before and 1 h after the injection of interferon. The management of depressive disorder in interferon therapy includes two different possibilities. Either interferon can be reduced to the lowest possible effective dose, although that often does not reduce depression sufficiently. The alternative would be a medical symptom suppression. This can be reached by cotreatment with different antidepressants. Selective serotonin reuptake inhibitors (SSRI), tricyclic antidepressants (TCA), and mirtazapine can be equally effective depending on the individual patient’s tolerance and response [2]. Interestingly different antidepressant drugs are effective in suppression of pruritus that is often an agonizing symptom of CTCL [15]. These drugs include mirtazapine and amitriptyline. Therefore, we would first line recommend a depression management upon interferon therapy with mirtazapine due to its lowest side effect profile and its proven effectiveness for pruritus control in CTCL [12].
4.4 New Individualized Therapeutic Options in Clinical Trials
4.4.1 Anti-CCR4 Therapy
Related posts:
 Concept and Scientific Background of Personalized Medicine
Concept and Scientific Background of Personalized Medicine
 Targeted and Personalized Therapy for Nonmelanoma Skin Cancers
Targeted and Personalized Therapy for Nonmelanoma Skin Cancers
 Personalized Management of Atopic Dermatitis: Beyond Emollients and Topical Steroids
Personalized Management of Atopic Dermatitis: Beyond Emollients and Topical Steroids
 Melanoma: From Tumor-Specific Mutations to a New Molecular Taxonomy and Innovative Therapeutics
Melanoma: From Tumor-Specific Mutations to a New Molecular Taxonomy and Innovative Therapeutics
 Autoinflammatory Syndromes: Relevance to Inflammatory Skin Diseases and Personalized Medicine
Autoinflammatory Syndromes: Relevance to Inflammatory Skin Diseases and Personalized Medicine
 The Personalized Treatment for Urticaria
The Personalized Treatment for Urticaria
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


