Pemphigus vulgaris
Pemphigus vegetans
Pemphigus foliaceus
Pemphigus erythematosus: localized
Fogo selvagem: endemic
Paraneoplastic pemphigus
Drug-induced pemphigus
IgA pemphigus
Pathogenesis
Pemphigus Target Antigens are Desmogleins
The hallmark of pemphigus is the finding of IgG autoantibodies against the cell surface of keratinocytes. The pemphigus autoantibodies found in patients’ sera play a primary pathogenic role in inducing blisters. When IgG fraction from patients is passively transferred to neonatal mice, the mice develop blisters with typical histologic findings [3]. Even monovalent Fab’ fragments of IgG or single chain fragment variables (monoclonal antibodies isolated using phage display technique, consisting of the variable regions of light chain and heavy chain of immunoglobulin) from patients are sufficient to cause blisters in neonatal mice, indicating complement activation and surface cross-linking may not be relevant in keratinocyte detachment [8–10].
Immunoelectron microscopy localized both pemphigus vulgaris and pemphigus foliaceus antigens to the desmosomes, the most prominent cell–cell adhesion junctions in stratified squamous epithelia [11]. Immunochemical characterization of pemphigus antigens by immunoprecipitation or immunoblotting with extracts from cultured keratinocytes or epidermis demonstrated that the pemphigus vulgaris and foliaceus antigens were 130 kD and 160 kD transmembrane glycoproteins, respectively [4, 5, 12]. The 160 kD protein recognized by pemphigus foliaceus sera was subsequently shown to be identical with desmoglein 1 (Dsg1) by comparative immunochemical studies [13].
Molecular cloning of cDNA encoding Dsg1 and pemphigus vulgaris antigens indicated that both molecules were desmogleins, which are the members of the cadherin supergene family [6, 7] (Fig. 33.1). Thus, pemphigus was discovered to be an anti-desmoglein autoimmune disease. The pemphigus vulgaris antigen was termed desmoglein 3 (Dsg3).
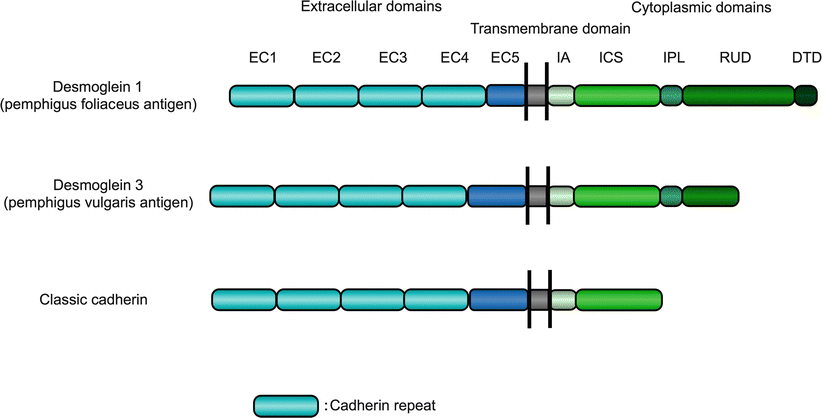
Fig. 33.1
Molecular structure of the pemphigus antigens. Cadherin is a single span transmembrane protein with a unique structure. The extracellular (EC) region of each cadherin member has four cadherin repeats of about 110 amino acid residues with calcium-binding motifs. Boxes with the same color have similarities in their amino acid sequences. Desmogleins have their own unique sequences of 29 ± 1 residues (repeating unit domain or RUD). ICS intracellular cadherin-specific domain, IA intracellular anchor domain, IPL intracellular proline-rich linker, DTD desmoglein-specific terminal domain
Desmosomes are intercellular adhesive junctions in the epidermis and mucous membranes and contain two major transmembrane components, desmogleins and desmocollins, both of which are cadherin type cell adhesion molecules. Desmogleins have four isoforms (Dsg1 to Dsg4). Expression of Dsg1 and Dsg3 is basically restricted to stratified squamous epithelia, where blisters are formed in pemphigus, while Dsg2 is expressed in all desmosome-possessing tissues, including simple epithelia and myocardium [14]. Dsg4 plays an important adhesive role mainly in hair follicles because mutations in DSG4 gene cause abnormal hair development [15]. The molecular structure of desmogleins is unique and they have four cadherin repeats in their extracellular domain as do classic cadherins and have an extra carboxyl-terminal domain containing repeats of a 29 ± 1 residues (Fig. 33.1).
Compelling evidence has accumulated that IgG autoantibodies against Dsg1 and Dsg3 are pathogenic and play a primary role in inducing the blister formation in pemphigus. Essentially, all patients with pemphigus have IgG autoantibodies against Dsg1 and/or Dsg3, depending on the subtype of pemphigus [16, 17]. When anti-desmoglein IgG autoantibodies are removed from patients’ sera of pemphigus vulgaris, pemphigus foliaceus or paraneoplastic pemphigus by immunoadsorption with recombinant desmoglein proteins, the sera are no longer pathogenic in blister formation [18, 19]. Furthermore, anti-desmoglein IgG autoantibodies that were affinity-purified from pemphigus sera on the desmoglein recombinant proteins can cause blisters when injected in neonatal mice [19, 20]. Some pemphigus sera react with Dsg4 due to cross-reactivity of a subset of anti-Dsg1 IgG, although Dsg4/Dsg1-cross-reacting IgG has no demonstrable pathogenic effect [21]. IgG autoantibodies against acetylcholine receptors or annexin-like molecules are reported, but their pathogenic relevance in pemphigus remains to be determined [22–24].
Thus, the basic pathophysiology of pemphigus is that IgG autoantibodies raised against Dsg1 and/or Dsg3 inhibit their adhesive function and lead to the loss of the cell–cell adhesion of keratinocytes, resulting in blister formation. Recent studies, in which pathogenic and non-pathogenic monoclonal antibodies were isolated from pemphigus patients, suggest that patients would have polyclonal IgG with diverse pathogenic activities [10, 25, 26]. Although the mechanism of blister formation in pemphigus is not fully understood, it is considered that the loss of cell-cell adhesion is triggered by the combination of direct inhibition of Dsg interactions (steric hindrance), the activation of cellular signal pathways, and endocytosis of cell surface Dsg [27–29].
Desmoglein Compensation Theory as Explanation for Localization of Blisters
Although the disruption of desmoglein-dependent cell adhesion by autoantibodies is the basic pathophysiology underlying blister formation in pemphigus, the clinical spectrum is more complex. The complex clinical features of pemphigus are explained logically by the desmoglein compensation theory: Dsg1 and Dsg3 compensate for each other when they are coexpressed in the same cell [30–32] (Fig. 33.2).
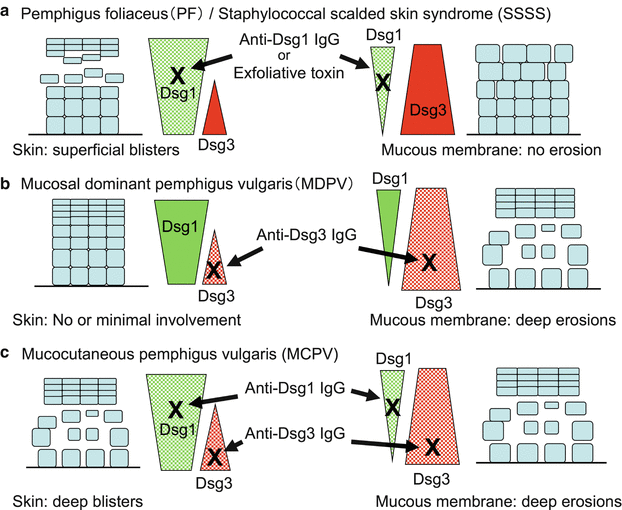
Fig. 33.2
Desmoglein compensation theory. Anti-Dsg1 IgG autoantibodies in serum from patients with pemphigus foliaceus or exfoliative toxin in staphylococcal scalded-skin syndrome (SSSS) cause superficial blisters in the skin; no blisters form in the lower epidermis or mucous membrane, because Dsg3 maintains cell-cell adhesion in those areas (a). Serum from patients with mucosal-dominant pemphigus vulgaris contains only anti-Dsg3 IgG, which causes mucosal blisters and erosions where there is no significant compensation by Dsg1, but no or minimal involvement in the skin where Dsg1 maintains cell-cell adhesion (b). Serum from patients with mucocutaneous pemphigus vulgaris containing anti-Dsg1 and Dsg3 causes blisters and erosions in the epidermis as well as in the mucous membranes because of no compensation (c)
The intraepithelial expression pattern of Dsg1 and Dsg3 is different between the skin and mucous membranes. In the skin, Dsg1 is expressed throughout the epidermis, but more intensely in the superficial layers, while Dsg3 is expressed in the lower portion of the epidermis, primarily in the basal and parabasal layers. In contrast, Dsg1 and Dsg3 are expressed throughout the squamous layer of mucosa, but Dsg1 is expressed at a much lower level than Dsg3.
Patients with pemphigus foliaceus have only anti-Dsg1 IgG autoantibodies (Table 33.2). Patients with the mucosal dominant type of pemphigus vulgaris have only anti-Dsg3 IgG autoantibodies, whereas those with the mucocutaneous type of pemphigus vulgaris have both anti-Dsg3 and anti-Dsg1 IgG autoantibodies [33, 34].
Table 33.2
Target antigens in pemphigus
Diseases | Autoantibodies | Antigens |
|---|---|---|
Pemphigus vulgaris | ||
Mucosal dominant type | IgG | Desmoglein 3 |
Mucocutaneous type | IgG | Desmoglein 3 |
Desmoglein 1 | ||
Pemphigus foliaceus | IgG | Desmoglein 1 |
Paraneoplastic pemphigus | IgG | Desmoglein 3 |
Desmoglein 1 | ||
Plectin/HD1 (500 kD) | ||
Desmoplakin I (250 kD) | ||
Desmoplakin II (210 kD) | ||
BPAG1 (230 kD) | ||
Envoplakin (210 kD) | ||
Periplakin (190 kD) | ||
alpha-2-macroglobulin-like-1 (170 kD) | ||
Drug-induced pemphigus | IgG | Desmoglein 3 |
Desmoglein 1 | ||
IgA pemphigus | ||
SPD type | IgA | Desmocollin 1 |
IEN type | IgA | ? |
For example, when sera contain only anti-Dsg1 IgG, which interferes with the function of Dsg1, the presence of Dsg3 compensates for the loss of function of Dsg1 in the lower epidermis. In contrast, in upper epidermis, there is no compensation by Dsg3. Therefore, blisters only appear in the superficial epidermis of the skin because that is the only area in which Dsg1 is present without coexpression of Dsg3. Although the anti-Dsg1 IgG binds to mucosa, no blisters are formed because of the coexpression of Dsg 3. Thus, sera containing only anti-Dsg1 IgG cause superficial blisters in the skin without mucosal involvement, as is seen in patients with pemphigus foliaceus.
The desmoglein compensation theory also explains the clinical and histological phenotype of bullous impetigo and staphylococcal scalded skin syndrome (SSSS) [32]. The blisters of bullous impetigo and SSSS are caused by exfoliative toxin (ET), which is produced by Staphylococcus aureus. ET was recently discovered to bind specifically to Dsg1 and cleave only the site after glutamic acid residue 381 between extracellular domains 3 and 4 [35–37]. When ET reaches the skin and digests Dsg1 in the lower layers of the epidermis, Dsg3 compensates for the loss of function of Dsg1 and manages to maintain cell–cell adhesion, while no compensation by Dsg3 occurs in the superficial layers of the epidermis. Therefore, ET induces superficial blisters on the skin. In mucous membranes, the Dsg3 expressed throughout the squamous layers of the mucosa compensates for the impaired Dsg1 and maintains cell–cell adhesion with no mucosal involvement.
Paraneoplastic Pemphigus Has a More Complex Autoimmune Reaction Than Classic Pemphigus
In addition to IgG autoantibodies against Dsg3 and/or Dsg1, patients with paraneoplastic pemphigus (PNP) develop characteristic IgG autoantibodies against multiple antigens with molecular weights of 500, 250, 230, 210, 190 and 170 kDa [19, 38] (Table 33.2). By immunochemical studies and cDNA cloning, most of these antigens were identified. The 500 kD antigen is plectin. The 250 kD and 210 kD antigens are desmoplakins I and II, respectively. The 230 kDa antigen is bullous pemphigoid antigen 1, the major plaque protein of the epidermal hemidesmosome and also a target antigen in bullous pemphigoid. The 210 kDa band also contains envoplakin. The 190 kDa antigen is periplakin, and 170 kDa antigen was recently identified as alpha-2-macroglobulin-like-1, a broad-range protease inhibitor [39, 40].
Anti-desmoglein antibodies play a role in inducing the loss of cellular adhesion of keratinocytes and initiate blister formation, while the pathophysiological relevance of the anti-plakin autoantibodies is unclear. The intracellular location of plakin proteins makes it unlikely that anti-plakin autoantibodies initiate pathology in paraneoplastic pemphigus because IgG cannot penetrate cell membranes. It is also important to bear in mind that not only humoral immunity but also cell-mediated cytotoxicity is involved in the pathogenesis of paraneoplastic pemphigus in a form of interface dermatitis. Patients with PNP show more severe and refractory oral erosions and stomatitis as well as more polymorphic skin eruptions when compared with classic forms of pemphigus. Recent studies using the pemphigus mouse model indicated that CD4+ T cells recognizing Dsg3 are able to directly infiltrate to dermal-epidermal junctions and induce interface dermatitis, suggesting the involvement of the cellular autoimmune reaction to epidermal antigens in PNP [41].
Clinical Features
Pemphigus Vulgaris
Pemphigus vulgaris has two clinical subtypes: mucosal dominant type and mucocutaneous type. Patients with mucosal dominant type show mucosal erosions mainly in the oral cavity with minimal or limited skin involvement. Patients with mucocutaneous type show extensive flaccid blisters and erosions on the skin in addition to the mucosal erosions. Any stratified squamous epithelia where Dsg1 and/or Dsg3 are expressed can be involved in pemphigus vulgaris.
Mucous membrane lesions are usually seen as painful erosions (Fig. 33.3). Intact blisters are rare, probably because they are fragile and break easily. Scattered and often extensive erosions may be seen on any part of the oral cavity. Extensive erosions and painful lesions in the mouth may result in decreased food and drink intake. Involvement of throat may produce hoarseness and difficulty in swallowing. The esophagus, conjunctiva, nasal mucosa, vagina, penis, anus and labia may also be involved. The diagnosis of pemphigus vulgaris tends to be delayed in patients presenting with only oral involvement, as compared to patients with skin lesions.
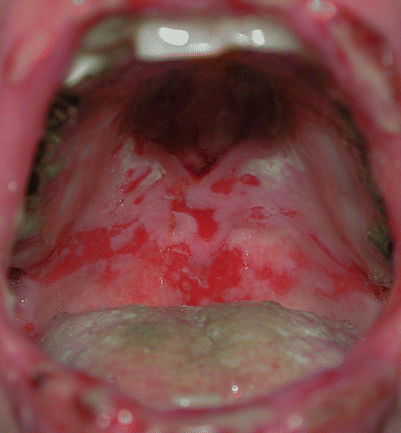
Fig. 33.3
Pemphigus vulgaris. Essentially all patients develop painful oral mucous membrane erosions
The primary skin lesions of pemphigus vulgaris are flaccid, thin-walled, easily ruptured blisters that appear anywhere on the skin surface (Fig. 33.4). The blisters arise on normal-appearing skin or erythematous bases. The blisters are fragile and soon rupture to form painful erosions that ooze and bleed easily. The erosions soon become partially covered with crusts that have little or no tendency to heal. Without appropriate treatment, pemphigus vulgaris can be fatal because large area of the skin lose epidermal barrier function, leading to loss of body fluids or secondary bacterial infection. Because of absence of cohesion in the epidermis, the upper layers are easily made to slip laterally by slight pressure or rubbing in active patients with pemphigus (Nikolsky sign).
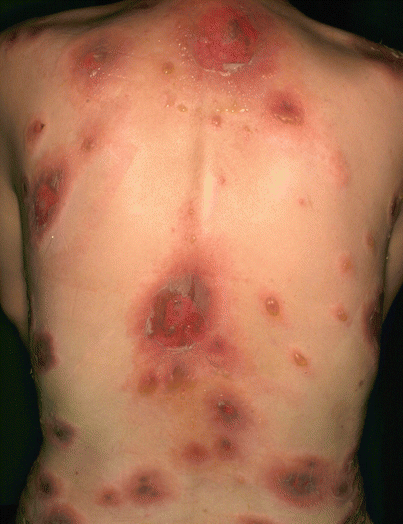
Fig. 33.4
Pemphigus vulgaris. Skin lesions are flaccid blisters which are fragile and soon rupture to form painful erosions that ooze and bleed easily
Pemphigus Foliaceus
Patients with pemphigus foliaceus develop scaly, crusted erosions, often on an erythematous base in the skin, but do not have apparent mucous membrane involvement even with widespread disease (Fig. 33.5). The absence of oral involvement may be a clue to clinically differentiate pemphigus foliaceus from pemphigus vulgaris.
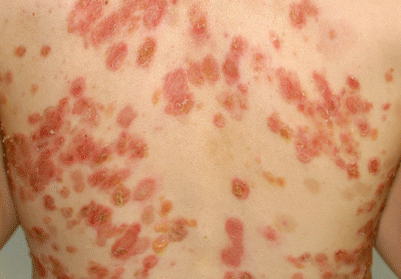
Fig. 33.5
Pemphigus foliaceus. Skin lesions are scaly crusted erosions and vesicles that are fragile and easily ruptured
The onset of disease is often subtle with a few scattered crusted lesions which come and go, and are frequently mistaken for impetigo. These lesions are usually well demarcated and scattered in a seborrheic distribution, including face, scalp, and upper trunk. Because the vesicle is so superficial and fragile, often only the crust and scale that result from a ruptured vesicle are seen. Disease may stay localized for years or may rapidly progress, in some cases, to generalized involvement resulting in an erythrodermic exfoliative dermatitis. Nikolsky sign is present. Generally patients with pemphigus foliaceus are not severely ill.
Paraneoplastic Pemphigus
Paraneoplastic pemphigus is a recently described form of pemphigus that occurs in association with underlying neoplasms [38, 39]. PNP is unique and distinct from the classic forms of pemphigus vulgaris and foliaceus by clinical, histologic, and immunopathologic criteria. The associated neoplasms are non-Hodgkin’s lymphoma (42 %), chronic lymphocytic leukemia (29 %), Castleman’s tumor (10 %), malignant and benign thymoma (6 %), spindle cell neoplasms (reticulum cell sarcoma) (6 %), and Waldenstrom’s macroglobulinemia (6 %). The combination of non-Hodgkin’s lymphoma and chronic lymphocytic leukemia represents almost two thirds of cases. Castleman’s disease, which is a very rare lymphoproliferative lesion, is the third most commonly associated neoplasm. The absence of common tumors, such as adenocarcinoma of breast or bowel and squamous cell carcinomas, is notable.
The most constant clinical feature of paraneoplastic pemphigus is the presence of intractable stomatitis (Fig. 33.6). The severe stomatitis is usually the earliest presenting sign and after treatment it is the one that persists and is extremely resistant to therapy. This stomatitis consists of erosions and ulcerations that affect all surfaces of the oropharynx and characteristically extend onto the vermillion of the lip. Most patients also have a severe pseudomembranous conjunctivitis with scarring. Esophageal, nasopharyngeal, vaginal, labial, and penile mucosal lesions may also be affected.
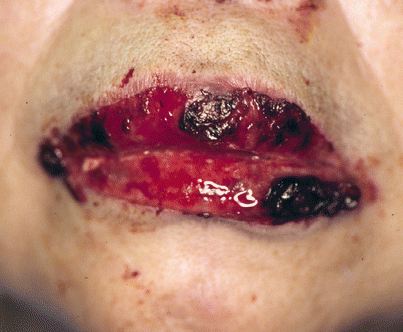
Fig. 33.6
Paraneoplastic pemphigus. The characteristic clinical feature is severe intractable stomatitis that extends onto the vermillion of the lip
The cutaneous lesions are quite polymorphic and may appear as erythematous macules, flaccid blisters and erosions resembling pemphigus vulgaris, tense blisters resembling bullous pemphigoid, erythema multiforme-like lesions, and lichenoid eruptions. Extensive cases show clinical resemblance with toxic epidermal necrolysis (TEN). The occurrence of blisters and erythema multiforme-like lesions on the palms and soles can be used to clinically differentiate paraneoplastic pemphigus from pemphigus vulgaris. Cutaneous lichenoid eruptions are very common together with severe stomatitis. In the chronic form of the disease lichenoid eruption may predominate over blistering lesions.
Paraneoplastic pemphigus is the only form of pemphigus that has involvement of non-stratified squamous epithelia. Approximately, 30–40 % of patients develop pulmonary symptoms [42, 43]. The earliest symptoms are progressive dyspnea, and pulmonary function studies show airflow obstruction, involving large and small airways, as seen in bronchiolitis obliterans, which can be fatal through respiratory failure. Recently, it has been demonstrated that the ectopic expression of Dsg3 or other epidermal antigens in the lung in the form of squamous metaplasia, which is often found in the lungs of PNP patients, is able to render the lung a target organ in PNP [44].
Other Forms of Pemphigus
Pemphigus Vegetans
Pemphigus vegetans is a rare vegetative variant of pemphigus vulgaris and considered to be one reactive pattern of the skin to autoimmune insult of pemphigus vulgaris. Pemphigus vegetans is characterized by flaccid blisters that become erosions and form fungoid vegetations or papillomatous proliferations, especially in intertriginous area and in the scalp or on the face. Pustules rather than vesicles characterize early lesions but these soon progress to vegetative plaques.
Pemphigus Erythematosus (Senear-Usher Syndrome)
Pemphigus erythematosus is simply a localized variant of pemphigus foliaceus. Typical scaly and crusted lesions of pemphigus foliaceus occur across the malar area of the face and in other seborrheic areas. Originally, pemphigus erythematosus was introduced for patients with immunological features of both lupus erythematosus and pemphigus, i.e. in vivo IgG and C3 deposition on keratinocyte cell surfaces as well as basement membrane zone and circulating antinuclear antibodies [45]. However, only few patients have been reported to actually have the two diseases concurrently [46].
Drug-Induced Pemphigus
There are sporadic cases of pemphigus in association with the use of drugs, such as penicillamine and captopril [47]. Pemphigus foliaceus is more common than pemphigus vulgaris in penicillamine-treated patients. Although most of patients with drug-induced pemphigus are shown to have autoantibodies against Dsg1 or Dsg3 [48], evidence suggests that some drugs may induce acantholysis without production of antibodies. Both penicillamine and captopril contain sulfhydryl groups that are speculated to interact with the sulfhydryl groups in desmoglein 1 and 3. Most, but not all, patients with drug-induced pemphigus go into remission after the offending drug is stopped.
IgA Pemphigus
IgA pemphigus is a newly characterized group of autoimmune intraepidermal blistering diseases presenting with a vesiculopustular eruption, neutrophilic infiltration, and in vivo-bound and circulating IgA autoantibodies against the keratinocyte cell surface, but no IgG autoantibodies [49–51]. IgA deposition on cell surfaces of the epidermis is present in all cases by direct immunofluorescence, and many patients have detectable circulating IgA autoantibodies by indirect immunofluorescence. There have been two distinct types of IgA pemphigus, subcorneal pustular dermatosis (SPD) type and intraepidermal neutrophilic (IEN) type. IgA autoantibodies in the SPD type react with desmocollin 1, which is expressed on COS7 cells [50], while autoimmune targets of the IEN type remain to be identified (Table 33.2). Subsets of IgA pemphigus patients have IgA autoantibodies against Dsg1 or Dsg3, making the autoimmune target of IgA pemphigus more heterogeneous. The exact pathogenic role of IgA autoantibodies in pustular formation in IgA pemphigus remains to be elucidated.
The patients with both types of IgA pemphigus clinically present with flaccid vesicles or pustules both on erythematous or normal skin. In both types the pustules tend to coalesce to form an annular or circinate pattern with crusts in the central area, although sunflower-like configuration of pustules is a characteristic sign of the IEN type. The predilection sites are the axillary and groin areas, but the trunk, proximal extremities, and lower aspect of the abdomen are commonly involved. Mucous membrane involvement is rare. Pruritus is often a significant symptom. Because the SPD type of IgA pemphigus is clinically and histologically indistinguishable from classic subcorneal pustular dermatosis (Sneddon-Wilkinson disease), immunological characterization is essential to differentiate the two diseases.
Histology
Pemphigus Vulgaris
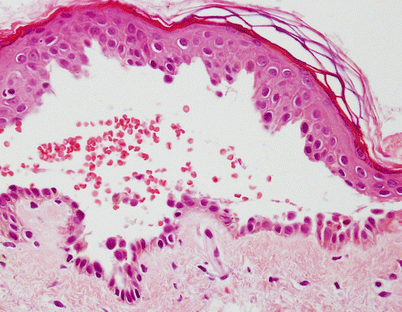
The characteristic histological finding of the classic form of pemphigus is intraepidermal blister formation due to loss of cell-to-cell adhesion (acantholysis) of keratinocytes without keratinocyte necrosis. In pemphigus vulgaris, acantholysis usually occurs just above the basal cell layer (suprabasilar acantholysis) (Fig. 33.7). A few rounded up (acantholytic) keratinocytes as well as clusters of epidermal cells are often seen in the blister cavity. Although the basal cells lose contact with their neighbors, they maintain their attachment to the basement membrane, thus giving the appearance of a “row of tombstones”. Eosinophilic spongiosis can be also seen in very early lesions of pemphigus.