CHAPTER 3 Pathophysiology of Varicose Veins
The World Health Organization defines varicose veins as ‘saccular dilatation of the veins which are often tortuous’.1 Further, this definition specifically excludes any tortuous veins associated with previous thrombophlebitis or arteriovenous connections or with venectasia.
Histochemical Physiology of Varicose Veins
Varicose veins differ from nonvaricose veins in physiologic function. This may occur in one or all of the histologic layers. Endothelial damage can occur in parts of a varicose vein,2 and has been noted both ultrastructurally and physiologically by a reduction in endothelial-mediated enhancement of norepinephrine (noradrenaline) induced vasoconstriction (Fig. 3.1).3,4
Characterization of the endothelin receptors in varicose veins compared with those in normal veins has shown decreased contraction to endothelin-1 in both varicose and saphenous veins of patients with primary varicosities. It may be that this observation will be associated with a decrease in the number of receptors.5
In most investigations the muscular layer has been found to be altered, with varicose veins having a considerable degree of smooth muscle hypertrophy and a 15% increase in muscle content compared with normal veins.6 This is thought to be a secondary response to venous hypertension. Other investigators have found that smooth muscle cells are capable of phagocytosis and decomposition of collagen fibers.7 Smooth muscle cells from varicose veins have been found to be less differentiated compared to normal veins and demonstrate increased synthetic capacity, greater proliferation and increased migration than smooth muscle cells found in normal veins.8 Thus, these cells may be part of the cellular basis for collagen breakdown. However, other investigators have noted a decrease in lactate dehydrogenase and creatine kinase activity in varicose versus normal veins and postulate that varicose vein weakness is due to a thinning or damaged muscular layer.9 This has been confirmed in a study of aging canine and human veins where a decrease in sympathetic innervation has been correlated with muscular layer thinning.10 In addition, the protein content of varicose veins (which is predominantly smooth muscle) is reduced.4 However, one research group has found no significant difference in the quantity of smooth muscle between normal and varicose veins.11
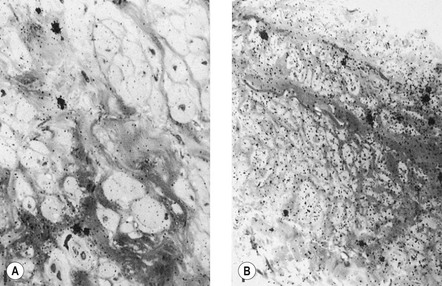
The adventitial layer has been noted to be altered in varicose veins. Some investigators have found that varicose veins have an extremely dense and compact fibrosis between the intima and adventitia, with a diminished and atrophied elastic network and a disorganized muscular layer (Fig. 3.2).12–15 Thickening and fibrillation of individual collagen fibers has also been noted.2,13,16,17 This translates to a reduced compliance that may lead to poor coaptation of venous valves and increased varicose vein wall stiffness. An in vivo measurement of venous elasticity in patients with normal, ‘high-risk’, and varicose veins confirmed reduced elasticity in both varicose and high-risk veins.18 In this study, individuals with high-risk veins were defined as having a family history of varicose veins, standing occupations, symptoms of venous disease and Doppler ultrasound reflux.
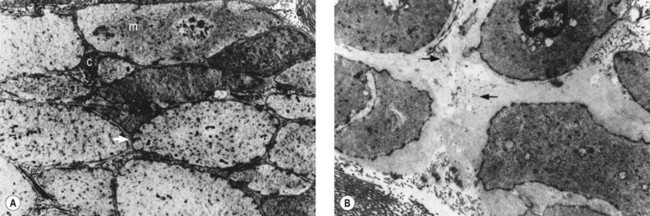
The described loss of tonicity of varicose veins is primarily the result of the loss of coordinated communication between smooth muscle cells. Electron microscopic studies of nonvaricose veins demonstrate the close approximation of smooth muscle cells. When veins become varicose, smooth muscle cells become vacuolated and are separated by collagen.12,13 With increasing varicose changes, intercellular collagen deposition accumulates and separates the smooth muscle cells, which then atrophy (Fig. 3.3). It is suggested that the resulting separation of smooth muscle cell hemidesmosomes causes inefficient smooth muscle contraction and increased venous distensibility.11,13,19 However, some varicose veins are capable of constricting in response to an infusion of dihydroergotamine. This venoconstriction is even more pronounced than that occurring in normal veins.20 The reason for this paradoxical effect is unknown, but a varicose vein appears to be a dysplastic vein characterized by malformations. Whether this is the result of continual high venous pressure or whether it is the primary etiologic event in the development of valvular incompetence is also unknown.
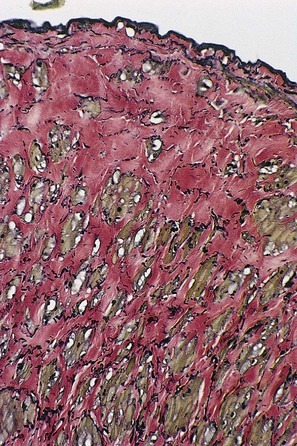
Elastin and collagen are known to play an important role in maintaining structural integrity of blood vessel walls. Normally when the wall is stretched, elastin generates a shortening force that opposes the traction exerted by the side branches and perivascular connective tissue and the lengthening force caused by pressure in the lumen. Type I collagen is believed to confer tensile strength to the vessel wall, whereas type III collagen may be involved in extensibility. In dilated and morphologically normal segments of varicose veins, type I collagen is present in a greater amount than type III. Furthermore, varicose veins contain more type I and type III collagen than do normal veins. It has been found that the elastin content is significantly reduced in dilated segments of varicose veins when compared with both normal veins and normal segments of varicose veins. Microscopically, the ratio of collagen to elastin appears to be significantly increased in the dilated segments of varicose veins. These findings tend to emphasize the important role of elastin in providing a retractile force that opposes development of dilation and tortuosity of the vein wall.21
Although collagen accumulation is thought to separate smooth muscle cells within the varicose vein wall, the collagen content of varicose veins is less than that in normal veins.6,22 The bulk of the varicose vein wall is made up of mucopolysaccharides and other ground substances. Varicose veins contain 67% more hexosamine (which comprises about 0.3% of normal vein dry material) than is found in normal veins.
Dysplasticity of the varicose vein wall may explain why varicose veins have an even greater susceptibility to pressure-induced distension than do nonvaricose veins. This anatomical–pathophysiologic correlation has been demonstrated by pharmacologic studies that show reduced maximal contraction of varicose veins compared with control veins.2,19 They have also been investigated with in vitro techniques measuring distensibility as a function of infused volumes of saline.23 However, some investigations have failed to discover a significant difference in the degree of intimal fibrosis between varicose and nonvaricose veins.24 Therefore, fibrosis of the vein wall alone is not totally responsible for the development of varicose veins.
In studying smooth muscle reactivity, the three main vasoconstrictor agents – norepinephrine, angiotensin II, and endothelin-1 – were compared. In diseased vein segments, a significant reduction in response to angiotensin II and norepinephrine was seen. Also, it was noted that there was reduction in response to endothelin-1. The reduction in angiotensin II affinity appeared at an early stage of varicose disease and supports the hypothesis that such an abnormality within the venous wall could play a role in the pathogenesis of primary varicose veins.25
Finally, a decrease in tocopherol concentration has been noted in varicose veins.26 A significant correlation also seems to exist between the inhibition of vessel wall tissue lipoperoxidation and their tocopherol concentration, independent of serum concentrations. This may be the result of the protective effect of blocking peroxidation of membrane-associated fatty acids by tocopherol and other antioxidants to prevent vein wall damage.27 It is clear that the dysplasticity of varicose veins correlates with the changes in their pharmacodynamics and histochemistry. Varicose veins have a demonstrated loss of contractility.28
Varicose veins are often complicated by local inflammation and thrombosis. This may be due to venous hypertension to an inherent histochemical abnormality in the varicose vein/endothelial wall. The formation of arachidonic acid-derived prostanoids was investigated in segments of varicose and nonvaricose veins. Venous production of prostacyclin was decreased, while that of thromboxane A2 and prostaglandin E2 was increased, in the varicose vein segments, regardless of whether they were macroscopically affected or unaffected.29 It is unknown whether this change in the cyclooxygenase pathway in the varicose vein wall is the cause or effect of its dysplasticity. In addition, histochemical examination discloses a marked increase in the activity of lysosomal enzymes,30 acid phosphatase, β-glucuronidase, and anaerobic isoenzymes (lactodehydrogenase) in primary varicose veins.31–33 These enzyme patterns suggest a decline in energy metabolism and an increase in cellular damage in the varicose veins. It has also been found that varicose veins accumulate and metabolize norepinephrine less efficiently than normal veins.34 Differences in expression and, probably more important, microscopic localization of matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinases between normal and varicose veins may explain the variability of disease between vein segments.35 MMP-2 has been found to cause relaxation of contracted vein segments which could lead to progressive venous dilatation, varicose vein formation and chronic venous insufficiency.36 Whether the abnormal level and/or action of MMP is the contributing factor or whether protracted increases in venous pressure lead to an increase in MMP expression is unknown.37 Therefore, both anatomical and biochemical abnormalities in the varicose vein wall contribute to its increased distensibility (Box 3.1).
Pathophysiology
Approximately 75% of the body’s total blood volume is contained within the peripheral venous system.38 The quantity of blood within the legs is a function of body position. When erect, 300 to 800 mL of extracellular and vascular fluids (the quantity varies according to the experimental method and the size of the subject measured) collects in the legs.39–41 This includes a 15% increase of blood volume.39 Thus, the venous system, especially in the legs, is an important component of the cardiovascular system’s circulatory reservoir. However, the arterial system plays an equally important role in cardiovascular adaptation to postural changes by virtue of changes in arterial resistance. In fact, studies have demonstrated that reflex changes in venous tone are not essential for this fluid shift.42
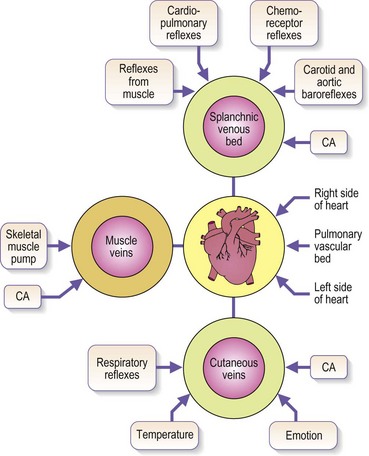
Venous blood pressure is determined by several factors. Among these are pressure generated by the heart, energy lost in the peripheral resistance of arterioles, hydrostatic gravitational forces, blood volume, anatomical composition of the venous wall, efficiency of one-way valves, vein wall distensibility (determined by hormonal, systemic alcohol and other factors), and contraction of venous smooth muscle as influenced by ambient temperature and sympathetic and parasympathetic nerve tone (Fig. 3.4).
Although arterial pressure is one factor in the development of venous pressure, arterial hypertension has been noted to be associated with the development of varicose veins in some epidemiologic studies43 but not others.44 Curiously, atherosclerotic disease has been linked epidemiologically to varicose veins, although it might be two common conditions occurring concurrently.45,46 It is postulated that this coincidence may be related to an atherogenic risk profile, owing primarily to coexistent inactivity, obesity and hypertension.46 At rest, in the erect position, pressure in the saphenous vein is determined primarily by the height of the column of blood from the right atrium to the site of measurement (90 to 120 mmHg at the ankle) (Fig. 3.5).47,48 Contraction of calf muscles generates pressures of between 200 and 300 mm Hg.49–51
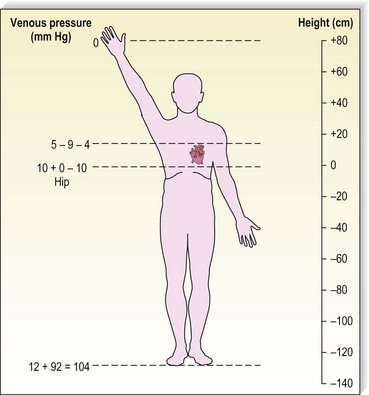
Figure 3.5 Venous pressure is that exerted by a column of blood from the heart to the location of measurement.
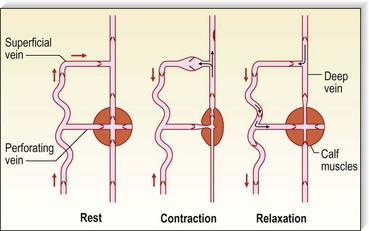
Pressure generated deep to the fascia, outside of muscles, is between 100 and 150 mmHg;51,52 however, with muscular activity, pressure in the normal saphenous vein at the level of the malleoli falls 45 to 68 mmHg below the resting level.53 It is reduced from 80 to 40 mmHg in the posterior tibial vein.54 Because of the one-way valves, blood flow is directed from the superficial venous system to the deep venous system through perforating vessels (see Fig. 1.4). This has been demonstrated visually by serial phlebography of the normal lower leg.55 The venous blood then flows towards the heart.
The venous pump in the foot is an important portion of the muscle pump of the lower leg. Weight bearing is usually necessary to propel blood up the leg. Bidirectional ultrasound velocity detector tracings of venous blood flow through the popliteal vein have demonstrated the importance of dorsiflexion of the foot when there is no weight bearing.56 Therefore, full flexion of the foot is important after sclerotherapy to maximize the efficacy of the lower extremity muscle pump.
Respiration produces alterations in intra-abdominal venous pressure. This ‘abdominal venous pump’ contributes to the flow of blood even when an individual is erect.41,57 Inspiration produces a rise in venous pressure in the external iliac vein, common iliac vein and inferior vena cava when measured in both the horizontal and erect positions (6.3 mmHg and 8.7 mmHg, respectively).57
In the supine position, blood flows evenly along all superficial and deep vessels towards the heart. It is propelled by the relatively small vis-à-tergo (force from behind) from the capillaries54 and the respiration-induced aspiration of blood into the abdominal and thoracic veins. In contrast to deep veins, superficial veins have smooth muscle in their walls. This allows contraction of these vessels in response to cold and to drugs such as dihydroergotamine58,59 and allows dilation in response to topical and systemic alcohol, estrogen and light physical trauma.54 As previously described, part of the pathophysiology of varicose veins may be a diminished response of such smooth muscle contraction.
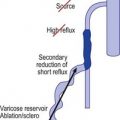
Regardless of its cause, chronic venous hypertension in the lower extremities causes an increase in venous diameter. This may lead to valvular insufficiency, which usually causes a reversal of blood flow from the deep veins into the superficial veins through incompetent perforating veins. This ‘private circulation’ may account for as much as 20% to 25% of the total femoral flow involved in a circular retrograde flow (Fig. 3.6).60,61
It has been found that prevalence of reflux in vein segments is correlated with signs of venous insufficiency, but in the general population, approximately 12% of limbs with no disease have reflux as detected by duplex ultrasound.62
Venous insufficiency has been correlated with standing occupations. A study that compared symptom-free vascular surgeons with normal individuals of nonmedical vocations showed that the superficial system was by far the most common site of venous incompetence in both groups. Vascular surgeons (standing occupation) showed a greater incidence of reflux than the controls. This was true even in subgroups in which reflux was seen in the superficial veins, as well as in those with reflux in the deep veins and perforating veins.63 In patients with symptoms of venous insufficiency, reflux in the great saphenous vein (GSV) territory is found in 85% of limbs, but only 68% of limbs show true saphenofemoral junction (SFJ) incompetence. Reflux is found in 20% of such patients in the small saphenous vein (SSV) territory, and strictly nonsaphenous origin of varicosities is found in 6%.64 One study demonstrated that 93% of the 10% of patients with nonsaphenous reflux in the study group were women with a mean of 3.2 pregnancies.65 This implied an association with female sex, hormones and/or number of pregnancies. Studies of causation of reflux focus on venous valves and vein walls. On one hand, an antiproteolytic milieu may favor the deposition of collagen and allow varicosities to develop;66 on the other hand, activation of monocytes and conversion to macrophages may cause weakening or destruction of valve segments.67
Direction of venous flow in varicose veins has been examined by McPheeters and Rice68 using fluoroscopy. Reversal of flow caused by incompetent perforator valves is beneficial during sclerotherapy. When a superficial varicosity is injected, its venous flow is forced distally to the smaller branching veins, where it is arrested (see Chapter 8).68 Thromboembolic disease is thereby prevented.
Superficial veins respond to increased pressure by dilating. Valvular incompetence occurs and varicosities appear.69 In addition, in muscular contraction, high compartmental pressure that normally occurs within the calf muscle pump is transmitted directly to the superficial veins and subcutaneous tissues drained by perforating veins.70,71 When this occurs, venous pressure in the cuticular venules may reach 100 mmHg in the erect position.54 This causes venular dilation over a broad area and may cause capillary dilation, increased permeability72–75 and a subsequent increase in the subcutaneous capillary bed through angiogenesis.73,76 This is expressed clinically as telangiectasia (venous blemishes). Histologically, cutaneous and subcutaneous hemosiderin deposition may also occur. This, in time, causes cutaneous pigmentation (see Chapter 2). However, some patients with chronic venous insufficiency are able to increase their venous blood flow through exercise.77 It is postulated that various factors (e.g. sympathetic tone, temperature, tissue metabolites) compensate venous hypertension to normalize cuticular blood flow. This finding demonstrates the complexity of the superficial venous system.
A special situation develops in the area of the medial malleolus. In this area, perforating veins are not surrounded by deep or superficial fascia. Therefore, any increase in deep venous pressure is transmitted directly through perforating veins to superficial connecting veins. This causes high cutaneous venous pressures and a transudation of extracellular fluid. This, in turn, leads to perivascular fibrin deposition, which has been blamed for decreased oxygenation of cutaneous and supporting tissues; this was thought to contribute to cutaneous ulceration (see Chapter 2).73,78,79 This theory has largely been discredited; the ability of any fibrin screen to prevent oxygen diffusion has never been proven.
The effect of temperature variations on the venous system is well studied.80,81 The cutaneous vasculature is intimately involved in thermoregulation. An increase in body core temperature results in cutaneous vasodilation. This does not occur as a result of relaxation of venous smooth muscle but because of the reduction in the vasoconstrictor impulses to the vein wall. Such vasodilation also occurs in varicose veins. Recently, strain gauge venous occlusion plethysmography has shown an increase in venous distensibility associated with temperature elevation.82 Similarly, alcohol ingestion may influence the development of varicose veins. Alcohol intake, like increased environmental temperature, causes cutaneous vasodilation. In an examination of 136 men with primary varicose veins greater than 4 mm in diameter, it was found that a significantly increased incidence of varicose veins occurred among men who consumed 4 oz (around 120 mL) of alcohol a day.83 Unfortunately, further experimental evaluations of this association have not been performed.
In summary, pathologic development of varicose veins can be divided into four broad categories, which may overlap and contribute to each other: increased deep venous pressure, primary valvular incompetence, secondary valvular incompetence and hereditary factors (such as vein wall weakness). All of these categories coexist and are influenced by temperature, alcohol, and hormonal and other vasodilatory stimuli (Box 3.2).
Increased Deep Venous Pressure
Most veins of the forearm and lower extremity remain competent even after maneuvers that induce venodilation and increase in blood flow, such as exercise hyperemia or postocclusion reactive hyperemia. However, veins with an inherent valvular weakness can be identified by reactive hyperemia in association with duplex flow analysis.84 The presence of femoropopliteal reflux is associated with clinical symptoms – it has been found in up to 15% of limbs having primary varicose veins. This is divided into those with superficial femoral venous reflux alone and those with isolated popliteal venous reflux.85
Proximal origin
Pelvic obstruction
Pelvic obstruction is an uncommon cause of varicose veins. Iliac vein compression syndrome is the phenomenon of compression of the left iliac vein by the left iliac artery overlying the fifth lumbar vertebra.86–90 This usually occurs in women, in whom it may be a cause of vulvar varicosities, but it has also been noted in men (see Chapter 5). Extravascular abdominal tumors, such as ovarian and uterine carcinoma or teratoma, may be causes of obstruction. More commonly, however, it is relative pelvic obstruction that provides a mechanism for impedance of return blood flow. Relative obstruction may occur in the third trimester of pregnancy, particularly during recumbency when the gravid uterus compresses the inferior vena cava against the lumbar spine and/or psoas muscles. Phlebographic studies have shown complete obstruction of the inferior vena cava at the confluence of the iliac veins in third-trimester pregnancies.91 Partial obstruction has been shown in earlier months of pregnancy. Some degree of compression is evident using phlebography, even in the left lateral decubitus position.
Increased intra-abdominal pressure
One popular hypothesis for the development of varicose veins is Western dietary and defecation habits that cause an increase in intra-abdominal pressure. A distended cecum or sigmoid colon, the result of constipation, may drag on the iliac veins and obstruct venous return from the legs.92 Population studies have demonstrated that a high-fiber diet is evacuated within an average of 35 hours.93 In contrast, a low-fiber diet has an average transit time of 77 hours. An intermediate diet has a stool transit time of 47 hours.
It is possible that the small increase in abdominal intravenous pressure caused by less than optimal bowel habits, when transmitted intravenously distally, gradually breaks down venous valves of leg veins.94,95 Evidence in support of this hypothesis is seen in populations of people who eat unprocessed high-fiber food. These persons are free of constipation and varicose veins.96,97 However, if this population’s diet is changed to low fiber, the incidence of varicose veins increases.92,98–101 In a diet that is intermediate between Western low-fiber and high-fiber diets, the prevalence of varicose veins is also found to be in an intermediate range.102
Defecatory straining induced by Western-style toilet seats has also been cited as a cause of varicose veins, in contrast to the African custom of squatting during defecation.99 However, venous pressures of subjects measured in both the sitting and squatting positions during defecation have not shown a significant difference.103 Venous flow has not been examined accurately in constipated and nonconstipated populations. Finally, there are other dietary factors besides fiber content that may explain the differences in prevalence of varicose veins. An increased incidence of varicose veins is found in populations of people who consume diets high in long-chain fatty acids, as opposed to diets high in short-chain fatty acids.104 Long-chain fatty acids have been shown in experimental systems to enhance blood coagulation and stimulate the development of blood clots.105–107 Clot lysis times were slower in the population group that consumed long-chain fatty acids.104 Accordingly, the type of dietary fatty acids consumed also may predispose the development of varicose veins. In addition, the Western diet has been found to be relatively deficient in vitamin E.108 It is hypothesized that the slight vitamin E deficiency, when aggravated by pregnancy, may predispose the vein wall to coagulation and fibrinolysis, thus causing the veins to become more sensitive to venous stasis and venous hypertension. Therefore, although it would seem prudent to recommend a high-fiber diet for several medical reasons, it remains an unproven treatment for the prevention of varicose veins.
An association between prostatic hypertrophy, inguinal hernia and varicose veins may be caused by straining at micturition with a resultant increase in intra-abdominal pressure.109
Another mechanism for increasing distal venous pressure by proximal obstruction is the practice of wearing girdles or tight-fitting clothing. A statistically significant excess of varicose veins was noted in women who wore corsets compared with women who wore less constrictive garments,110 although this finding was not confirmed by a subsequent study.111 However, a more recent study of 20 women aged 20 to 46, who wore ‘tight’ jeans (degree of compression was not measured), found that in 14 of these women there was an increase in subcutaneous pressure from 10 to 15 mmHg at rest to 30 mmHg when walking, as opposed to no change in pressure when wearing loose-fitting clothing. 112 Thus, the use of tight jeans can negatively affect venous return. A similarly increased incidence was noted in women who stand at work compared with those whose jobs entail more walking and sitting.45,47,110–116 However, this has not been confirmed universally.117,118
Leg crossing and sitting on chairs are two other potential mechanisms for producing a relative impedance in venous return. Habitual leg crossing is commonly thought to result in extravenous compression, but this has never been scientifically verified. A decreased incidence of varicose veins has been noted in population groups that do not sit in chairs.119,120 It is thought that sitting may produce some compression on the posterior thigh which produces a relative impedance to blood flow. Wright and Osborn121 have shown that the linear velocity of venous flow in the lower limbs in the recumbent position is reduced by half in the standing position and by two-thirds when sitting. Alexander120 found that the circumferential stress on the saphenous vein at the ankle was 2.54 times greater with chair sitting than with ground sitting. This may explain the increased incidence of varicose veins in men versus women in population groups in which only men sit on chairs and women sit on the floor. In this population study,119 varicose veins were present in 5.1% of men and only 0.1% of women. Finally, the practical implications regarding chair sitting concern those who travel for long periods in airplanes. Pulmonary thromboembolism has occurred in many people after prolonged air travel and has been termed economy class syndrome.122 Although pre-existing venous disease, dehydration and immobility are all contributing factors, chair sitting adds another insult to the venous system.
Most,43–45,115,116,123–129 but not all,111,125,130,131 studies have found that obesity is associated with the development of varicose veins. Careful examination of some of these epidemiologic studies shows that when the patient’s age is correlated with obesity, the statistical significance is eliminated.132 Obesity was especially correlated with the development of varicose veins in women when the varicosities occurred in unison with cutaneous changes indicative of venous stasis (see Chapter 2).129,133,134,135 This may be secondary to decreased exercise and associated medical problems specific to obesity, such as hypertension, diabetes, hypercholesterolemia and sensory impairment.136
Running has been demonstrated to raise the intra-abdominal pressure by 22 mmHg.137 This increase in abdominal pressure occurs because of a reflex tightening of abdominal muscles during running, which prevents the pelvis from tipping forwards during thigh flexion induced by contraction of the iliopsoas muscle group.138 Therefore, during strenuous leg exercise, elevated abdominal pressures may impede venous return. By way of comparison, a Valsalva maneuver was shown to elevate the intra-abdominal pressure by 50 mmHg or more.137 Strenuous exercise, particularly long-distance running, is often associated with prolonged increases in limb blood flow, which theoretically could overload the venous system and lead to progressive dilation.69 Usually, dilated veins that occur in this situation are normal and do not require treatment. Finally, it is commonly noted that occupations that require standing for prolonged periods have an increased incidence of varicose veins.139 This may be exacerbated by tall height, although this factor has not been supported by other studies.132
Saphenofemoral incompetence
Saphenofemoral impedance is rarely caused by anatomical abnormalities in the saphenofemoral triangle. When it occurs, pelvic tributary veins or accessory saphenous veins may converge in such a manner that flow to the femoral vein is impeded.140 Likewise, iliac venous incompetence caused by the congenital absence of venous valves, or by damage to the valves through thrombosis, may cause distal venous hypertension.
Our interest and focus on the venous valve dysfunction as a fundamental cause of distal venous hypertension began with unpublished observations using angioscopy. The angioscope provided a direct view of the internal architecture of saphenous veins. Patients taken to surgery who demonstrated preoperative reflux verified by duplex ultrasound showed a variety of pathologic lesions in the valves themselves. The first indication was a relative paucity of valves. The observation of a decrease in the number of GSV valves had already been reported by Cotton in 1961.141 Next, we encountered actual valve lesions. These observations were an extension of those reported by Hoshino et al,142 who classified valve damage in the saphenous vein into three categories ranging from stretched commissures to perforations and valve splitting.
From the preceding observations we suggest that the earliest valve defect is an increase in the commissural space, which allows reflux on the border of the vein. This may be one of the earliest causes of reflux in varicose veins. Later, thinning, elongation, stretching, splitting and tearing of the valves develop. The last stages are thickening, contraction, and possibly even adhesion between valves. These observations have been confirmed by Van Cleef et al.143 While we have proposed that this valve damage is acquired and causes axial reflux as well as outflow through check valves in perforating veins, others have proposed that the cause of primary venous insufficiency is an actual low number of valves in the saphenous system.144
The angioscopic observations could be confirmed by gross morphologic studies that, when extended to microscopy observations using monoclonal antibody labeling, have demonstrated monocytic infiltration into damaged venous valves.145 Others have found leukocytic infiltration into varicose veins and have called attention to the fact that the cells observed released vasoactive substances, including histamine, tryptase, prostaglandins, leukotrienes and cytokines.146 Observations in patients led to the conclusions that venous hypertension was related to leukocytic infiltration on the cranial surfaces of the venous valve and venous wall and that leukocytes there were greater in quantity than on the caudal portion of the valve leaflets and venous wall.
This inflammatory sequence occurred early during the phase of venous hypertension and progressed further after release of the occlusion. The model showed that venous occlusion with elevation of the hydrostatic pressure caused a highly injurious process for the surrounding tissues. It was accompanied by formation of microhemorrhages on the high-pressure side of the postcapillary venule and rolling and adhesion of leukocytes on the venular endothelium.147
Van Bemmelen et al148 produced a model of venous hypertension by creating arteriovenous fistulas in Wistar rats using microsurgical techniques. Valvular incompetence was seen as early as 1 day after creation of the arteriovenous fistula, and valvular structural changes were noticeable within 2 months of production of venous hypertension. Elongation of the cusps was observed. Separation and leakage of the cusps were encountered along the entire valvular free border, and, in later stages beyond 4 months, valve areas became difficult to recognize because commissures were lost and bulging of the valve sinus disappeared.
We have pursued this line of investigation and have reproduced the human observations in the animal model.149–153
Another model of venous hypertension has been produced by Lalka.154 This model creates venous hypertension by ligation of the inferior vena cava, the common iliac veins and the common femoral veins. This preparation elevates rat hindlimb venous pressures compared with forelimb pressures. Myeloperoxidase assay indicates leukocyte trapping in hindleg tissues in the same way as it occurs in humans.155
The observations just mentioned suggest that valve damage in venous insufficiency is an acquired phenomenon related to leukocyte and endothelial interactions and an inflammatory reaction. This observation is not universally accepted. A study on 13 valve structures from varicose GSV showed an absence of lymphomonocyte infiltration in 85% and rare isolated ‘nonsignificant’ inflammatory cells in 15%.156 However, if this hypothesis is correct, pharmacologic intervention to block leukocyte adhesion, activation and subsequent valve damage may be a possibility.
Distal origin
Valvular incompetence
Unlike that described in the previous section, incompetence of the SFJ clearly produces distal retrograde flow into the GSV and thus produces distal venous hypertension (Fig. 3.7). The GSV then dilates, producing further distal valvular incompetence sequentially. Retrograde flow thus produced is channeled through the perforator veins back into the deep venous system. This produces a private circuit of blood flow from the femoral vein to the saphenous vein and back to the femoral vein through perforating veins.60 It has been estimated that the total volume of flow in this circuitous route may be 20% to 25% of the total limb blood flow during exercise.61 This paradoxical circulation can be maintained for a long time, but eventually the quantity of blood channeled by the perforator veins increases. As this happens, there is hypertrophy and dilation of the superficial veins, producing valvular incompetence and localized varicose veins.
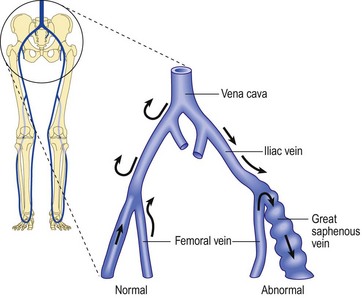
In addition to increasing superficial venous volume through perforator incompetence, retrograde flow produces an increase in acidity and potassium concentration with a decrease in venous oxygen concentration. These three factors promote vasodilatation to exacerbate venous stasis.157
Perforator incompetence in the lower part of the leg may occur from localized thrombosis in the vein following trauma. It is believed that localized thrombosis is usually masked by the local tissue injury. The valve cusps become involved by the thrombus and, after recanalization, remain functionless.158 Dodd and Cockett54 found, on examination of 54 legs with perforator incompetence, that the lower leg incompetent perforators were in communication with the soleal plexus of veins and, as such, were the channels most likely to be damaged as a result of thrombotic episodes in this region. In support of this concept, Fegan,159 Hobbs,160 Lofgren,161 and Beninson and Livingood162 have all pointed out that the treatment of varicosities may have no effect on superficial venous pressure. These investigators contend that treatment of perforating veins draining the ankle and lower calf area is important.70,110,160,163–165 The vessels may be either surgically ligated70,146–165 or sclerosed (see Chapters 9 and 10).160 Only then will retrograde flow under high pressure through the calf muscle pump be diverted upstream away from the skin. When this is done, a lowering of cuticular venous pressure and a decrease in dermal capillary pressure is accomplished. The excess transudation of fluid-producing edema and the associated decrease in tissue oxygenation and nutrition is halted. Quill and Fegan166 have demonstrated clearly the narrowing of the proximal SSV after obliteration of incompetent distal perforating veins in 9 out of 11 cases. This suggests that dilation and incompetence of the saphenous vein may be caused by distal reflux, as well as primary or irreversible abnormalities of the proximal venous wall. The finding has been confirmed through duplex examination of patients with cosmetic veins or primary varicose veins.
In a study of 500 lower limbs in ‘cosmetic’ patients, incompetence from below the knee extending upwards was found in 63.3%. However, only 9% were found to have perforator incompetence.167 An additional study of 167 consecutive patients with primary varicose veins demonstrated that 31% had incompetence of the GSV but no evidence of SFJ incompetence, and 24% of limbs had incompetence of the SSV without incompetence of the SFJ.168 Thus, perforator incompetence, although important as an etiologic factor, is not solely responsible for varicose vein development in all patients.
Duplex scanning has contributed to knowledge regarding valvular dysfunction produced by venous thrombosis. For the most part, deep valvular insufficiency comes from direct valve destruction rather than obstruction-induced venous pressure elevation and dilation of the vein wall. Destruction of the valve cusps occurs after venous thrombosis. It follows that the extent of venous valvular incompetence is related to the extent of the original deep venous thrombosis (DVT). One could extrapolate from these observations that valvular competence might be preserved if thrombus could be removed quickly by thrombolytic agents.169
Direct ambulatory venous pressure measurements and duplex examination have shown bidirectional flow through perforator veins. Exercise has been found to cause inward flow from the dilated superficial system and perforating veins into deep veins.170 The perforator vein here functions as a drainage pipe to limit cuticular venous hypertension. This explains the dilation of re-entry perforating veins, which drain the superficial reflux into the deep venous circulation and which become competent after superficial vein surgery.171–174 Compression distal to perforator veins in patients with venous disease demonstrated inward flow in 55 out of 56 perforator veins examined with duplex scanning.175 Thus, venous hypertension is related to both perforator incompetence and valvular dysfunction.


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


