© Springer International Publishing Switzerland 2015
Arin K. Greene, Sumner A. Slavin and Håkan Brorson (eds.)Lymphedema10.1007/978-3-319-14493-1_22. Pathophysiology of Lymphedema
(1)
Department of Surgery, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, Suite MRI 1005, New York, NY 10065, USA
Keywords
LymphedemaT-helper cellsT-helper 2 cellsObesityIL-13IL-4TGF-betaFibrosisAdipose tissue depositionInflammationKey Points
Lymphedema is a pathophysiologic process resulting from injury, infection, obstruction, or congenital defects in the lymphatic system.
Primary lymphedemas occur as a result of genetic or developmental abnormalities in the lymphatic system; secondary lymphedemas are caused by secondary insults to the lymphatic system
The histological hallmarks of lymphedema are lymphatic fluid stasis, chronic inflammation, fibroadipose tissue deposition, and hyperkeratosis.
Risk factors for development of lymphedema include genetic abnormalities, obesity, radiation, and infection.
Cellular mechanisms of lymphedema are unknown; however, recent studies have demonstrated a critical role for CD4+ cells in the regulation of fibrosis in animal models and correlative clinical studies.
Introduction
The lymphatic system is an essential component of the circulatory system whose main roles are maintaining fluid homeostasis, acting as a conduit for migration and transport of immune cells, regulation of inflammatory responses, and enabling dietary absorption of fat. Networks of lymphatic vessels begin as blind-ended lymphatic capillaries and transport interstitial fluid unidirectionally back to the heart. Disruption of lymphatic vasculature secondary to chronic parasitic infections, during the course of cancer treatment, after trauma, or as a consequence of genetic mutations results in stasis of protein rich interstitial fluid and chronic inflammation. These pathologic changes, over time, lead to lymphedema; a progressive disease characterized by adipose deposition and tissue fibrosis. It is well established that patients with lymphedema have significant impairments in quality of life, recurrent infections, and in some cases deadly secondary tumors.
Broadly speaking, lymphedema can be categorized as either primary or secondary. Primary lymphedema, as the name implies, is caused by abnormal development of the lymphatic system or pathological changes intrinsic to the lymphatic vasculature. These developmental abnormalities may be related to genetic defects that either directly or indirectly regulate lymphatic differentiation and function. In contrast, secondary lymphedemas occur as a consequence of postnatal iatrogenic, infectious, or traumatic insults to the lymphatic system. Although both primary and secondary lymphedemas share similar pathologic features including chronic swelling, inflammation, adipose deposition, and fibrosis, there important pathologic distinctions remain between patient responses, disease course, and response to treatments. In addition, although recent studies have improved our understanding of the pathology of lymphedema in general, the mechanisms that regulate these disease specific responses remain unknown and are an important area of research.
Classification of Lymphedema
Primary Lymphedema
Primary lymphedemas can be classified either by the timing of presentation, mode of inheritance (genetic linked or sporadic), or region of pathology (e.g., systemic or visceral). Traditionally, the timing of presentation has been used most commonly to categorize patients as having congenital lymphedema (i.e., present at birth), developing lymphedema around the time of puberty (lymphedema praecox), or presenting in adults older than 35 years (lymphedema tarda). The vast majority of patients present with either congenital lymphedema or lymphedema praecox; lymphedema tarda is diagnosed in less than 10 % of patients [1].
Classification of lymphedema based on timing of presentation is not particularly useful since there is no reference to the pathological causes. More recently, Connell et al. published a classification system and diagnostic algorithm that subcategorizes congenital lymphedemas as syndromic, systemic/visceral, disturbed growth, congenital onset, and late onset [2]. This classification system is helpful because it takes a more functional approach to lymphatic development and will likely aid in identifying genetic mutations due to its more inclusive nature.
Primary lymphedemas, in general, affect females twice as often as males and tend to more frequently involve the lower extremities. It is estimated that nearly 30 % of patients with primary lymphedema have an identifiable genetic mutation (often in the signaling pathway for vascular endothelial growth factor-C (VEGF-C)). The most studied example of this scenario is Milroy’s disease, a form of congenital primary lymphedema that is caused by a heterozygous inactivating mutation of FLT4, the gene that encodes for the receptor for VEGF-C (Vascular Endothelial Growth Factor Receptor-3 (VEGFR-3). Milroy’s disease is a familial, sex-linked condition and accounts for approximately 2 % of all lymphedemas. These patients most commonly have bilateral lower extremity lymphedema that is, in some cases, accompanied by hydroceles. Another common genetic cause of lymphedema is an autosomal dominant condition known as lymphedema–distichiasis syndrome. These patients have an autosomal dominant mutation in the FOXC2 gene, resulting in a combination of lower extremity lymphedema and an extra row of eyelashes.
Sporadic (i.e., not familial) cases are the most common causes of primary lymphedema accounting for ~60 % of all patients with lymphedema. The most common form of sporadic primary lymphedema is Meige’s disease. Patients with this disorder present with symptoms usually around the time of puberty with a female to male ratio of 4:1. These facts have led some authors to suggest that female sex hormones may play a role in the development of lymphedema.
Secondary Lymphedema
Secondary lymphedema is the most common cause of lymphedema and develops secondary to either direct or indirect injury to the lymphatic system. The most common form of secondary lymphedema worldwide is filariasis, a condition in which parasitic roundworms Wuchereria bancrofti occupy the lymphatic vasculature, thereby blocking the flow of lymph from the extremity. Although the true incidence of filariasis remains unknown, estimates ranging between 140 and 200 million are commonly cited among patients residing primarily in third world countries [3]. In contrast, this form of lymphedema is very rare in developed countries.
In developed countries secondary lymphedema occurs most commonly as a complication of cancer treatment with breast cancer being the most common cause. It is estimated that nearly one in three patients who undergo axillary lymph node dissection for staging or treatment of breast cancer go on to develop lymphedema. Lymphedema, however, is not limited to breast cancer survivors, as a recent study demonstrated that nearly one in six patients who undergo treatment for other solid tumors such as melanoma, sarcoma, and gynecological malignancies also go on to develop lymphedema [4]. Even relatively minor disruption of the lymphatic system with sentinel lymph node biopsy, a procedure in which only a few lymph nodes are sampled for cancer staging, can result in lymphedema in 5–7 % of patients [5]. Lymphedema can also develop in patients who do not have lymph node biopsy but rather wide skin excisions (for example during the course of treatment for sarcoma or melanoma) particularly when these procedures are combined with radiation therapy suggesting that extensive injury of either the deep (i.e., lymph nodes) or superficial (i.e., dermal) lymphatic system can result in lymphedema development.
Importantly, the development of lymphedema after lymphatic injury usually occurs in a delayed fashion. Thus, although virtually all patients experience minor swelling immediately following surgery, in the vast majority of cases the swelling resolves within the first 4–6 weeks. However, a subset of patients develop recurrent swelling at a later point, typically 6–12 months postoperatively (77 % within the first 3 years) that does not resolve. In these cases, the diagnosis of lymphedema can be made when other causes of swelling (e.g., recurrent disease, deep venous thrombosis, systemic fluid overload,) are ruled out. This diagnosis is often confirmed with physiologic studies such as lymphoscintigraphy or indocyanine green near infrared angiography demonstrating diminished lymphatic transport capacity, dermal back flow, and dysfunctional lymphatic vessels. Lymphedema may even develop after very prolonged periods of time in at-risk patients with the longest reported case occurring 30 years after the initial surgery. In these cases, often an inciting event such as an infection or additional injury precedes the development of progressive limb swelling and lymphedema.
Recent studies have suggested that progression of lymphedema may be retarded during its early stages through the use of conservative measures such as compression garments or manual lymphatic massage therapy. Although there is some debate regarding the efficacy or timing of these treatments in preventing lymphedema development, the fact that measures aiming to decrease interstitial fluid stasis can alter disease progression/development is interesting and suggests that lymph stasis (rather than lymphatic injury alone) is necessary for development of lymphedema. However, once lymphedema develops it is usually progressive and chronic in nature although there is wide variability in the rate at which pathologic changes occur. Thus, in some cases lymphedema has a smoldering course with relatively mild changes in limb volume or tissue changes over very long periods of time (often years), while in other cases there is rapid progression of disease with disabling swelling and physiologic changes.
Risk Factors for Lymphedema
A large number of epidemiologic studies have analyzed genetic and comorbid factors that increase the risk of developing lymphedema. A clear understanding of these risk factors is important in preoperative surgical consultation and can be used as a means of tailoring surgical procedures to individual patients in an effort to decrease the risk of lymphedema development.
Genetics
The discovery of lymphatic markers and mechanisms that regulate lymphangiogenesis has led to an attempt by numerous researchers to test the hypothesis that mutations in the coding or noncoding regions of these genes increase the risk of developing primary or secondary lymphedema. The majority of these studies have been performed in patients with primary lymphedema; however, recent studies have also targeted genetic risk factors for secondary lymphedema. The study of genetic risk factors for development of secondary lymphedema is interesting and based at least partially on the observation that some patients with breast cancer-related lymphedema exhibit abnormalities in lymphatic transport even in their unaffected normal extremity.
An interesting report by Mendola and colleagues from the lymphedema research group analyzed genetic mutations in 78 patients with familial (i.e., inherited) and 149 patients with sporadic primary lymphedemas. The investigators found that mutations in seven genes encoding molecules regulating VEGFR3 and its downstream pathways were responsible for 36 % of inheritable forms of primary lymphedema. In contrast, only 8 % of patients with sporadic primary lymphedema exhibited these mutations [6]. These findings are important and demonstrate that complex pathways regulate development of hereditary lymphedema. More importantly, these studies highlight the need for additional study since the majority of hereditary and sporadic primary lymphedema patients did not exhibit known genetic mutations.
Recent studies provide support for the theory that genetic mutations may also increase the risk of secondary lymphedema after surgery. For example, Feingold et al. sequenced the coding and flanking noncoding regions of the hepatocyte growth factor (HGF) and its high affinity receptor MET in 59 women with breast cancer-related upper extremity lymphedema, and 159 unrelated matched controls. This analysis was based on previous studies demonstrating that this signaling pathway is an important regulator of lymphangiogenesis in a number of physiologic settings [7]. Interestingly, the authors identified an increased rate of mutations in HGF/MET pathways in patients with lymphedema suggesting that impairments in lymphatic regeneration after injury due to dysfunctional HGF/MET signaling contributes to an increased risk of developing lymphedema. In a follow-up case–control study (80 patients with breast cancer-related lymphedema compared with 108 breast cancer controls), Feingold and colleagues found mutations in CJC2 (encoding connexin 47), a gap junction protein that is thought to regulate dermal lymphatic transfer, in four patients with lymphedema but not in any of the controls. Similar mutations have been observed in cohorts of patients with primary lymphedema [8]. Interestingly, in contrast to their previous report the authors identified only one patient with a MET mutation, suggesting that larger samples of patients are needed for these studies.
Newman and colleagues used a nested case–control approach to study genetic changes in 22 patients who developed breast cancer-related lymphedema within 18 months of surgery (case) as compared to 98 patients who did not develop lymphedema. The authors reported that single nucleotide polymorphisms (SNPs) within VEGFR2, VEGFR3, and RORC were associated with development of lymphedema [9]. SNPs are variations in DNA sequence that occur normally in the general population. These variations can occur in both coding and noncoding regions of the DNA and although gene function may be normal under physiologic conditions, some SNPs may lead to gene function changes that increase the risk of pathology. Future studies in this arena should focus on identifying the functional gene changes that occur in patients with SNPs that increase the risk of lymphedema after breast surgery.
Obesity
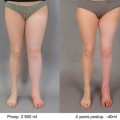
The increased risk of developing lymphedema in obese patients is well described in previous epidemiologic studies. In a seminal study in 1957, Treves followed over 1,000 patients after treatment for breast cancer and found that obese patients were at significantly increased risk of developing lymphedema [10]. This observation has been confirmed in numerous subsequent studies. For example, Werner et al. reviewed 282 patients with upper extremity lymphedema after treatment for breast cancer and showed that patients with a higher body mass index (BMI) also had a higher incidence of lymphedema. Another prospective study examined the risk of developing lymphedema in the upper extremity in 137 patients with breast cancer and showed that patients with a BMI >30 had a threefold greater risk than patients with a BMI < 25 [11]. The best supporting evidence is a randomized controlled trial in which patients with upper arm lymphedema underwent 12 weeks of dieting and nutritional counseling as compared with patients who did not. The results showed that patients who lost weight had significant reductions in arm volumes and upper arm lymphedema when compared to the control patients who did not diet [12]. These results suggest that lymphatic impairment in obesity is reversible. More importantly, these studies show that obesity and lymphedema are intricately linked. This is not surprising since a defining feature of chronic lymphedema is progressive adipose tissue deposition. This observation has led some authors to conclude that lymphedema may be a form of “regional” obesity in which tissues are more prone for depositing fat in the setting of caloric excess. This hypothesis is supported by the fact that lymphatic fluid has been shown to increase adipocyte proliferation and differentiation and by other studies demonstrating that even mild lymphatic injury activates expression of genes necessary for adipocyte activation.
Obesity, independent of surgery, has been shown to decrease lymphatic function. For example, previous studies have shown that obese patients have decreased clearance of macromolecules from the skin and subcutaneous tissues as compared with lean individuals [13, 14]. In addition, Greene and colleagues have shown that morbidly obese patients (BMI > 59) develop primary lower extremity lymphedema (i.e., without antecedent trauma or injury) characterized by decreased lymphatic transport and dermal back flow on lymphoscintigraphy [13




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
