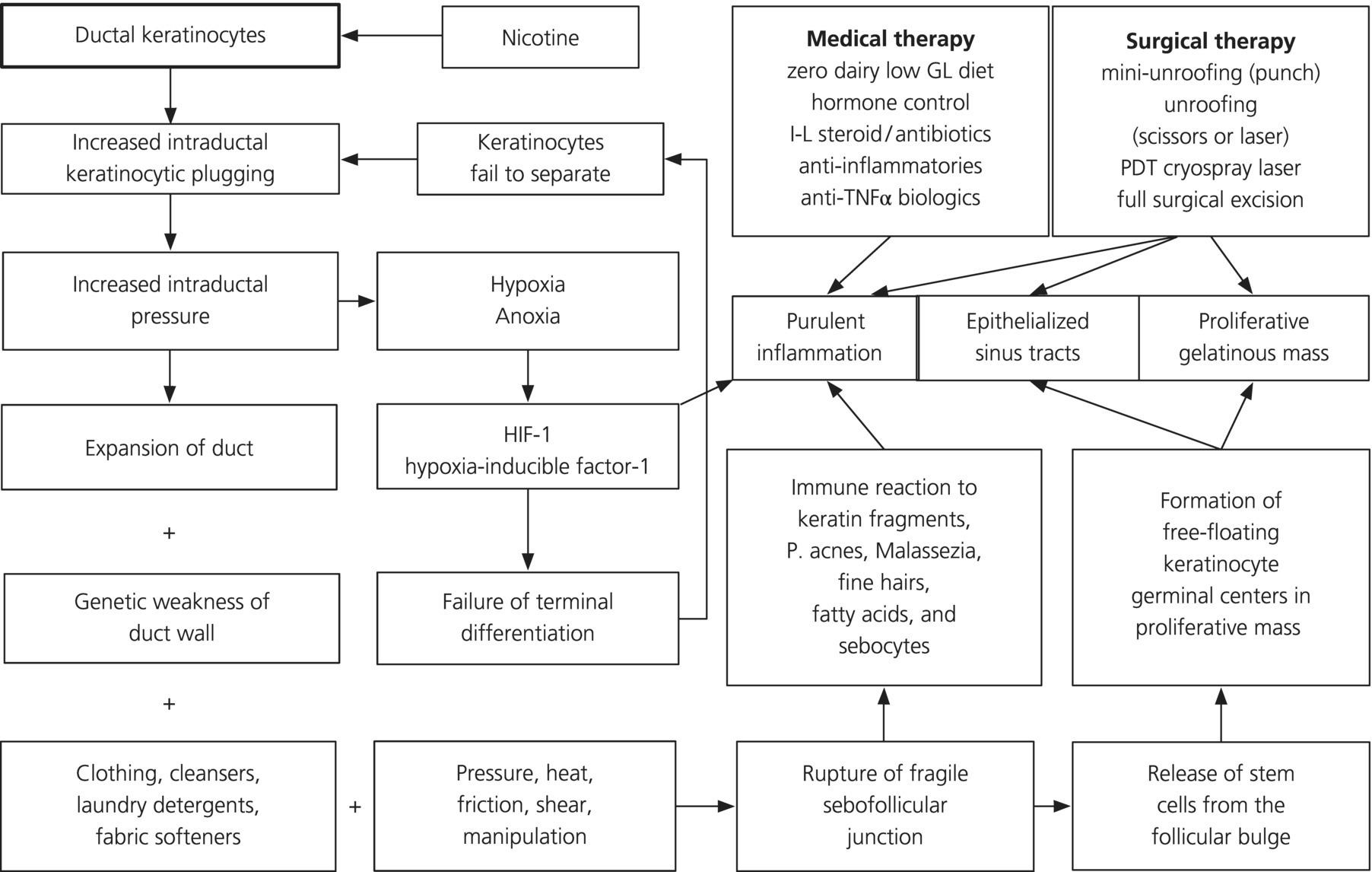
Chapter 3 The three acnes share several mechanisms that create their specific different appearances, but not all mechanisms are active in all three acnes. That accounts for their morphological variability, so that while plugging of the follicular portion of the folliculopilosebaceous unit (FPSU) is common to all, obvious comedo (visible blackhead and whitehead) development is variable and the ductal obstruction is not clinically visible in some acnes. In addition to the three primary acnes, there are several variants that illustrate exaggeration of one or more of the active pathogenic processes. (See Section 3.4, “Other Variants.”) For all readers, it is necessary for you to know that what follows is a synthesis. Much as I would prefer to write about a fully integrated and proven pathogenesis, science isn’t quite there yet. That being so, what you will read here is a mixture of facts and conjecture based on these facts. The challenge, to suggest ethical experiments that successfully test the hypotheses, is considerable. Hormones, especially the androgen dihydrotestosterone (DHT), drive the increased production of the keratinocytes that line the follicular portion of the folliculopilosebaceous unit (FPSU). As the lining cells accumulate and grow inwards, the mass created presses outwards against the walls of the follicle. This expansion leads to an increase in pressure within the follicle, and the increase in pressure leads to a lack of oxygen (hypoxia and eventually anoxia). The lack of oxygen encourages the growth of Propionibacterium acnes, a bacterium that grows well where there is no oxygen. Indeed, it is the increase in the population of this anaerobic or micro-aerophilic organism that is the marker that allowed the leap in comprehension linking the lack of oxygen (whether relative or absolute) to the slowing of terminal differentiation described in this chapter. The hypoxia and anoxia appear to have additional effects on keratinocyte metabolism and inflammatory response. (See Section 7.3.) At the same time as the follicular tube is filling up and plugging, the androgenic hormones are also driving the increased production of sebum by stimulating a marked increase in the production rate of sebocytes. The two major organisms living in the duct can break down the triglycerides (one of the fats in the sebum) into fatty acids for food. This provides nutrition to the P. acnes and also to the Malassezia yeast in the follicle. The hypothesis suggests that the lack of oxygen (and other nutrients) caused by the increased pressure impairs the ability of the follicular keratinocytes to undergo terminal differentiation. This is the process through which basal keratinocytes change over their life span from soggy little cuboidal basal cells filled with a metabolically active gel to a thin flat sheet made up mostly of keratin that simply flakes off into the duct at the end of its life. If maturation and differentiation are interrupted, the keratinocyte does not fully mature and does not desquamate. The lining keratinocytes remain attached to each other instead of separating (desquamating). That leads to the continued buildup of material in the follicle, and the plug in the pore becomes larger. At the same time, using electron microscopy, a buildup of lipid-containing vacuoles (little bubbles of lipid) can be seen in the stacked layers of keratinocytes that have stopped differentiating and are piling up in the follicular canal [1]. These lipids likely represent both the source of metabolic energy (that is not being used as energy because there is insufficient oxygen for oxidization) and an unused reserve of the lipid that was intended for incorporation into the lipoprotein end products. In this hypothesis, these lipids are not being used because their metabolism is shut down because of the anoxia. The androgenic-acnegenic hormones, if not controlled, continue to stimulate the basal cells of this closed follicular system from the outside, producing more and more internal pressure until the walls of the follicles leak or eventually rupture. This takes time, measured in months when young healthy follicles are the target, which is the usual case in teenage acne. Up to this point, the entire acne production process is due to hormonal stimuli. This is the stage of non-inflammatory acne. Basically, all there is to see at this point is a plugged and swollen follicular unit and a busy oil gland. The bacteria and yeast in the follicular canal, the intrafollicular flora, are just innocent bystanders, happy to be consuming the sebum (their preferred food) and multiplying actively. The leaks and the ruptures that occur at this point in the follicular wall release materials from the follicular canal, including bacteria and yeast and other materials contained within the follicle. These materials, including flakes of keratin in the case of acne inversa [2], leak into the tissue under the surface of the skin, in the upper and mid-dermis. That release of normally intrafollicular contents triggers both of our two immune systems. As these foreign materials interact with the innate immune system, they are termed stimuli. When the same materials interact with the adaptive (acquired) immune system, they are called antigens. The immune systems awaken, and the fight is on. The resulting inflammatory cascade lights the fires of inflammation, a molecular and cellular conflagration termed the inflammasome. The leaks in the walls of the follicles become a two-way street, allowing some of the inflammatory mediators access to the contents of the duct, leading to intrafollicular collections of inflammatory cells that push toward the surface as folliculopustules. Most of the inflammation remains outside the FPSU structure, especially in acne inversa. This severe acne variant is distinguished by its failure to discharge to the surface and its failure to subsequently heal quickly, as normally occurs with boils or abscesses due to infection. Beyond this point, inflammation rules and previously non-inflammatory acne becomes inflammatory, one FPSU at a time. As Leeming et al. state, It is probable … that when micro-organisms are present in acne lesions they make a significant contribution to the promotion of inflammation. Indeed it would be surprising if they did not, considering the large number of microbial antigens and metabolic by-products which would be released into the dermis from a colonized acne lesion when ruptured. [3] The complex interactions that occur have recently been elucidated by Melnik in a remarkable synthesis of the dozens of processes that mediate the inflammasome via Notch1. Acne inversa is the variant explored in his landmark paper, but the same processes are doubtless expressed in the less florid acnes [4]. The basic premise is that the rupture of the FPSU releases damage-associated molecular pattern molecules (DAMPs). These fragments bear molecular patterns that identify them as, for example, pieces of yeast or hair or virus. They are recognized as foreign by toll-like receptors (TLRs), and the TLRs turn on the numerous pro-inflammatory cytokines that manage the inflammatory reaction. This inflammation is normally downregulated by Notch signaling, so when Notch signaling is diet impaired, TLR-mediated inflammation is not held in check as it should be, permitting and therefore causing more inflammation. To add to the problem, Notch1 is also involved in regulation of keratinocyte differentiation. Lack of Notch1 leads to a reduced induction of p21, the earliest regulatory step in the process. Less Notch1 means less differentiation of the keratinocytes and so more ductal plugging. While control of the effects of excess hormonal stimulation is essential to contain the process and prevent new lesions from forming, hormonal control alone will not provide the anti-inflammatory action needed for an acceptable clinical response and resolution. The object in fighting this fire must be to shut off the fuel supply as well as get to work with fire extinguishers. That means that an essential part of therapy is the total elimination of the intrafollicular flora if possible. Reversing the whole pathogenetic process once it reaches this stage requires reversal of the hormones, reversal of the follicular plugging, elimination of the bacteria and yeast, and reversal of the inflammation. It is a tall order and not one that can be accomplished with monotherapy. A team attack is essential. The folliculopapular and folliculopustular forms of acne rosacea are started by the same hormonal stimuli that trigger acne vulgaris. Several local differences lead to a markedly different-looking disorder. First, the support of follicular walls of the FPSU is, I suspect but cannot prove, slowly weakened by years of exposure to ultraviolet-A (UVA) and UVB light, and the weakness is most prominent at the upper end of the follicular unit where the sun has been strongest and where actinically damaged collagen is commonly seen. (See Section 1.2.) This has two consequences. First, the distribution of acne rosacea is over the convex, sun-exposed parts of the face. Second, the rupture of the follicular wall, with consequent formation of the folliculopapule or folliculopustule, occurs without the development of a visible comedo. It is proposed that these visible plugs do not form because the follicular wall has been so compromised by photodamage from the sun that the leaks and ruptures occur before a visible plug can accumulate. Demodex might even find the UV-damaged and dilated follicular orifices more commodious and so an easier place to breed and raise their families. Second, the flora and fauna in acne rosacea are different from those in acne vulgaris. The hypothesis suggests that the pressure required to induce the apparent anoxia cannot occur because the follicular wall bursts too soon. It follows that there is none of the profound anoxia deep in the follicular duct as is found in acne vulgaris, so enhanced populations of P. acnes are not the important players here. Instead, the problem is inflammation induced by a combination of stimuli and antigens from retained vellus hairs, Demodex folliculorum, a smaller population of P. acnes, and the yeasts, mainly Malassezia but occasionally Candida species. Early acne rosacea may also differ from later acne rosacea because antibiotic therapy will change the bacterial and yeast proportions in the intrafollicular cavity. This is because the oral antibiotics, used as anti-inflammatories, reduce the population of P. acnes and other bacteria. At the same time, this enhances the population of Malassezia that is normally present as an innocent commensal on the face and permits (or encourages) other yeasts such as Candida. As with acne vulgaris, hormonal control alone will not provide the anti-inflammatory therapy needed for an acceptable clinical response in acne rosacea. The intrafollicular flora and fauna must be eliminated as an essential part of therapy. The addition of Demodex further complicates an already complex picture. Again, from Leeming, “It is conceivable that the causes of inflammation vary amongst lesions and individuals, and that this variation could explain apparent paradoxes such as the different responses of individual acne patients to various therapies” [3]. Leeming’s concept is further illustrated by what seems to be another process underway in acne rosacea when it is being treated with low-dose doxycycline. One of the early selling points used in promoting this low-dose approach was the claim that it was too low to encourage overgrowth of vaginal and vulvar candidiasis [5]. It is reasonable then to suggest that this induces less “biotropic” effect on the Malassezia organism as well, inducing Malassezia growth to a lesser degree and so tending to allow the cooling of the inflammation without inducing the variant of acne rosacea that requires ketoconazole for control. Third is the need to avoid sun damage. This is an obvious concern with regard to the damage to the collagenous support tissues of blood vessels as well as the follicles. Such actinic damage induces the actinic telangiectasia that usually accompanies the folliculopustular component of acne rosacea (although it often stands alone). The geographic coexistence of these two damaged tissues underlines the common etiological factor, sun damage. This link is an essential part of the hypothesis here. Thus, effective broad-spectrum sun protection from an early age is highly important to prevent and to minimize cumulative and further damage to both follicular support tissues and the superficial blood vessels in the area. Unfortunately, a major part of that battle is often lost by the time clinical disease occurs, leaving only prevention of further damage as an option. Physical rehabilitation of the vascular changes with several modalities of selectively destructive laser is possible, but is difficult, time-consuming, and expensive, and it does not return the dilated vessels to normal. The UV damage to the supporting fabric of the follicular canal that may cause marginal dilation (and thus provide the Demodex with a more accessible home) is a defect not directly amenable to laser or other repair. Nevertheless, resurfacing and other techniques can provide impressive cosmetic benefit using very selective surface destruction followed by re-epithelialization, and/or microsized scars followed by a healing “tightening effect.” The hormones that activate the other acnes are operative here as well, and their control is essential to slowing the plugging of the follicular portion of the duct. Again, there are regional differences in the way the lesions evolve. For one, visible open comedones are rare to nonexistent in the early stage of the disease. Comedones do show up as the massively distorted structures called multiheaded or tombstone comedones later in the disease, but these are end-stage structures, explained elsewhere in this volume. Obviously, however, there is something plugging the pores. Certainly, the hormonal stimulus is operative here and is extensively reviewed elsewhere. The problem is that the follicular plug is in the deeper portion of the follicular part of the FPSU, in the infrainfundibulum, and it is invisible from the surface unless profoundly filled with follicular keratinocytic debris. (See Figure 3.1.) Figure 3.1 The chain of events from keratinocyte excess to medical and surgical management of the plugged and exploded duct is simplified here. Excess keratinocyte production begets the early plug that expands and triggers the anoxia that, likely through hypoxia-inducible factor 1, interferes with terminal differentiation and induces inflammation. Building upon work in acne pioneered by the Burkharts [6], recent work in AI/HS points a finger at biofilm formation as a possible contributor [7]. Biofilm is a protective mechanism for intraductal organisms and is an additional attractive explanation for the plugging of the duct. By protecting the organism against contact with antibiotics and other threats, such a film may facilitate the persistence of P. acnes and other film-forming organisms in the follicular duct and thus contribute to the problem. The Staphylococcus and Streptococcus occasionally encountered in the area, and thought to be innocent, may play a role in this way, protecting themselves and other organisms in the process [8]. If one takes a close look at the general shape and the development of the AI/HS lesions, it is apparent that there has always been a clue to what is really happening under the skin in this disorder. The clue lies in the fact that, unlike acne vulgaris and acne rosacea pustules (and ordinary bacterial and Candida folliculitis), the follicular structure in AI/HS does not rupture and discharge vertically to the surface of the skin. Instead, it ruptures at a level deep in the dermis (Figure 3.2). These lesions do not “point” to the surface. They rupture horizontally and then both pyogenic (pus-causing) inflammation and a peculiar gelatinous proliferative mass infiltrate the subcutis, sometimes for significant distances, usually horizontally but to depth as well, along paths of least resistance.
Pathogenetic mechanisms summarized
3.1 Acne vulgaris
3.2 Acne rosacea
3.3 Acne inversa/hidradenitis suppurativa (AI/HS)
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


