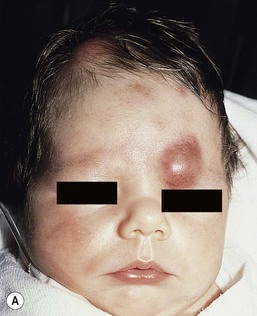
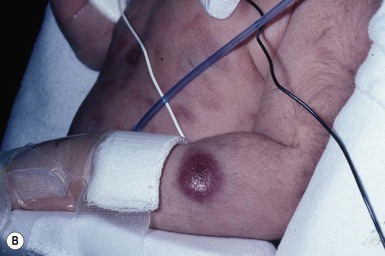
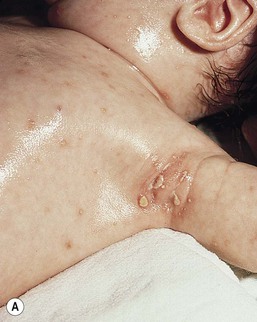
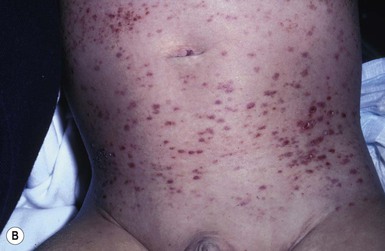
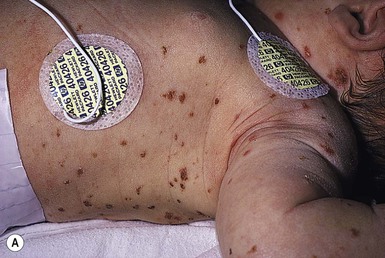
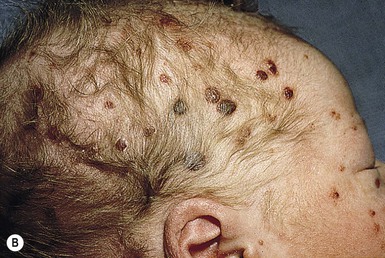
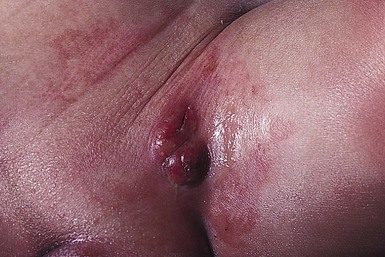
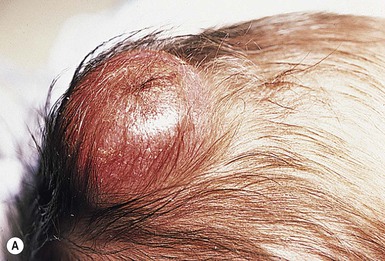
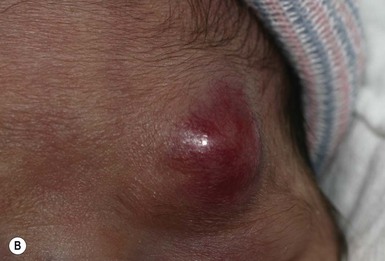
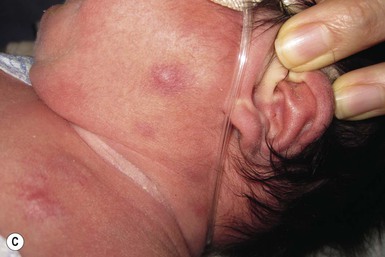
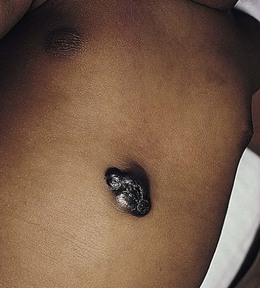
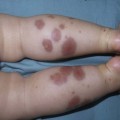
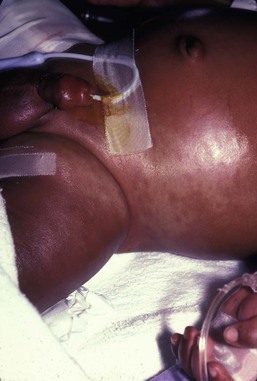
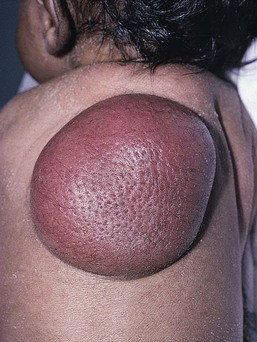
Neil S. Prose, Richard J. Antaya Skin disorders characterized by infiltrative lesions can be present at birth or develop during the first few months of life. Some represent frank neoplasms, both benign and malignant, whereas others are the result of metabolic errors. In most instances, diagnosis is facilitated by skin biopsy, in which certain cell types can be identified by special stains and immunologic markers. Others require special enzyme assays for definitive diagnosis.1 Congenital leukemia is a rare hematologic disorder. Less than 1% of all childhood leukemia is diagnosed in newborns.2 The majority of neonates have acute myeloid leukemia (AML), while acute lymphoblastic leukemia (ALL) is less common. The most frequently occurring chromosomal abnormality in both congenital AML and ALL is a translocation involving the mixed lineage leukemia (MLL) gene. To differentiate congenital leukemia from the several infectious and proliferative disorders that can easily mimic this condition, the following diagnostic criteria are employed: 1. The presence of immature white cells in the blood 2. Infiltration of these cells into extrahematopoietic tissues 4. The absence of chromosomal disorders that are associated with ‘unstable’ hematopoiesis (such as trisomy 21).3,4 The cutaneous manifestations of congenital leukemia consist of petechiae, ecchymoses, and skin nodules. Leukemia cutis is seen in approximately 60% of patients with congenital leukemia. The firm nodules are usually 1–2.5 cm in diameter and blue to purple in color (Fig. 28.1). They are often widely spread over the skin surface, although congenital leukemia may present as a single nodular cutaneous lesion.5 Diffuse calcinosis cutis in an infant with congenital AML has been reported.6 Congenital AML may be manifested by ‘blueberry muffin’ lesions,7 and Darier’s sign has been observed.8 The clinical signs and symptoms include hepatosplenomegaly, pallor, lethargy, and respiratory distress. Lymphadenopathy occurs in some infants. Biopsy of a cutaneous lesion reveals a dense pleomorphic mononuclear cell infiltrate in the dermis and subcutaneous fat. Atypical mitotic figures may be present. The diagnosis of congenital leukemia can usually be confirmed by a complete blood count, bone marrow aspirate, and radiographs of the skull and long bones. In every case, cytogenetic studies must be performed to exclude the possibility of transient myeloproliferative disorder, which is usually associated with trisomy 21.9 This disorder is also associated with a diffuse vesiculopustular eruption (see Chapter 10).10,11 Congenital leukemia is often fatal, with overall survival ranging from 10–25%.5 Treatment consists of intensive multiagent chemotherapy. The benefit of hematopoietic stem cell transplantation remains uncertain. Spontaneous remission of congenital leukemia cutis, usually without involvement of blood or bone marrow, has been reported.12–14 Leukemia occurring in infants under the age of 1 year bears many similarities to congenital and neonatal forms. Patients in this age group also have a high incidence of MLL rearrangements. Typical lesions include violaceous macules, nodules, and plaques. Patients with AML may present with larger purplish tumors, while those with ALL may develop numerous prurigo-like papules. The development of granulocytic sarcoma (formerly known as chloroma) in the periorbital skin of infants has also been reported.15 Langerhans’ cell histiocytosis (LCH), a rare proliferative disorder, may be present at birth or may develop during the first few months of life. The estimated incidence of neonatal LCH is 9/1 000 000 in infants <1 year of age, with disease presentation during the first month of life in about 6% of those affected.16,17 The spectrum of clinical presentations, and the clinical course, is extremely varied and ranges from the simple presence of one or several nodules, widespread crusted papules and vesicles, to severe, progressive multisystem disease. The most frequent cutaneous presentation – particularly in infants and newborns – consists of multiple widespread vesiculopustules with umbilication and a hemorrhagic crust.18–23 Lesions tend to favor the scalp, trunk, diaper area, and skin folds (Fig. 28.2A). They may begin as subtle brown or pink papules, and evolve into crusted or purpuric lesions (Fig. 28.2B). Papular lesions may coalesce into areas of superficial ulceration with oozing, especially in intertriginous areas. Characteristic lesions also include fissures behind the ears, and crusting and oozing of the external ear canals. The scalp is a frequent site of involvement, and the coalescence of crusted and scaling lesions may lead to partial alopecia. Presentation as a ‘blueberry muffin baby,’ with cutaneous hematopoiesis, has been reported.24 Nodules and petechiae, which may involve the palms and soles, are also observed. Oral lesions are relatively common, and can develop before skin lesions. They may appear as superficial ulcerations or erosions, but these are often associated with underlying alveolar bone disease. Other oral manifestations include gingival bleeding, facial swelling, and pain. Premature eruption and loss of teeth, and destruction of alveolar bone are characteristic. Involvement of the vulva and vagina may also occur. Nail involvement consists of subungual pustules, paronychia, onycholysis, and longitudinal grooving. Permanent nail dystrophy may result.25 A multifocal congenital form of LCH, which has sometimes been called ‘Hashimoto–Pritzker disease’26 is characterized by papulonodular and papulovesicular skin lesions. Cases presenting with a solitary nodule have also been reported. These solitary skin nodules are usually red-brown and often have a hemorrhagic quality, ulceration, or crusting.27 Most affected infants have lesions at the time of birth without evidence of other organ system involvement (Figs 28.3, 28.4).26 Skin lesions typically resolve spontaneously, without sequelae but localized areas of scar or atrophy can occur.28–30 However, skin relapse, bone disease, late-onset diabetes insipidus, and even progression to severe LCH has been reported in children who present with congenital skin lesions.31 The majority of infants with all forms of congenital or neonatal Langerhans’ cell histiocytosis will show evidence of multisystem disease.31–33 Bone lesions are the most frequent noncutaneous manifestation. Findings may include asymptomatic lytic lesions, deformation, fracture, or medullary compression. The skull is a common site, and disease in that location manifests on X-ray as punched-out lesions in the cranial vault. Mastoid involvement may lead to mastoid necrosis, and destruction of the ossicles may result in deafness. Intercranial disease may cause exophthalmos and diabetes insipidus. Lymph node involvement is seen in a significant percentage of patients, and tends to occur in the cervical chain. Pulmonary disease may be associated with cough and tachypnea. Other findings include bullae formation and diffuse interstitial fibrosis. Invasion of the liver may cause mild cholestasis, but eventually evolve to sclerosing cholangitis. Severe fibrosis of the liver results in ascites, jaundice, and liver failure. Involvement of the spleen may worsen the severity of thrombocytopenia. Gastrointestinal disease is reported to occur in a small percentage of patients.34 Characteristic findings are vomiting, diarrhea, protein-losing enteropathy, and failure to thrive secondary to malabsorption. The most common endocrine manifestation is diabetes insipidus. This occurs most often in children with extensive disease and involvement of the skull. Langerhans’ cells may also infiltrate the thyroid and pancreas. Biopsy of a cutaneous lesion reveals a diffuse infiltration of histiocytes with abundant eosinophilic cytoplasm and eccentric, indented nuclei. In some cases, the nuclei may appear pleomorphic or atypical. Positive CD1a and/or CD207 (Langerin) staining of the lesional cells is needed for a definitive diagnosis. Electron microscopy is no longer required since it is now known that the expression of Langerin correlates with the ultrastructural presence of Birbeck granules.35 Preliminary laboratory evaluation of the patient with biopsy-proven skin lesions must include a complete blood count (CBC), serum electrolytes, urine specific gravity, and assessment of liver function. Examination of the bone marrow may show the presence of increased histiocytes. Skeletal survey, chest radiographs, and magnetic resonance imaging (MRI) may be used to determine the extent of bone, pulmonary, liver, spleen, and CNS involvement. The pustular and intertriginous lesions of LCH in neonates and infants may be confused with candidiasis, seborrheic dermatitis, psoriasis and scabies. Congenital LCH must also be differentiated from other neoplastic disorders, such as leukemia and lymphoma; from congenital infections, especially herpes simplex; and from those viral disorders associated with ‘blueberry muffin’ lesions. The majority of children with infantile LCH develop multisystem disease. The prognosis seems to be better in congenital ‘self-healing’ LCH, but multisystem disease is still possible, and such neonates must be followed to assure that they do not develop extracutaneous disease later on. Survival of infantile LCH in the past few decades has improved from 57% to 74%.16,35 Single-system disease has a significantly higher rate of survival (94%), but patients with initial liver involvement have a much poorer prognosis, with a 25% 5-year survival. In children with LCH limited to the skin (<5% of cases), a ‘wait and see’ approach should be accompanied by careful monitoring for disease progression. Children in this category may benefit from therapy with mild to moderate-strength topical corticosteroids. Children with multisystem disease are treated with chemotherapy; standard treatment is based on steroids and vinblastine. Mycosis fungoides is extremely rare in young children, but a number of cases have been well documented.36 The patients presented with widespread and persistent scaly plaques. Several cases were initially misdiagnosed as either atopic dermatitis or tinea corporis, leading to a significant delay in diagnosis. This form of cutaneous T-cell lymphoma is characterized by large tumor cells that express CD30 antigen. Clinical presentations consists of papules, and plaques which may ulcerate. The rare occurrence of this disease during the first 2 years of life has been documented.37 Lymphomatoid papulosis is a chronic, recurrent disorder characterized by the presence of reddish brown papules and nodules. Progression to malignant forms of lymphoma is rarely reported. In one series of pediatric patients, including children under the age of 2 years, a benign course, accompanied by a predominance of CD8+ lymphocytes was observed.38 Primary cutaneous B-cell lymphoma is extremely rare in infancy. Primary cutaneous lymphoblastic lymphoma, presenting with firm violaceous nodules with overlying telangiectasia, has been reported to occur in some children under the age of 2 years.39,40 Subcutaneous panniculitis-like T-cell lymphoma, with purplish tender nodules in an acral distribution, has also been reported to occur in infancy.41,42 Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening condition characterized by hepatosplenomegaly, cytopenias and prolonged fever.43 Central nervous system (CNS) symptoms are frequent. Some children have been noted to have an evanescent macular and papular skin eruption, sometimes associated with episodes of fever (Fig. 28.5). Genetic HLH occurs in familial forms (FHLH), and in association with a variety of immune deficiencies.44 Acquired and genetic forms may both be triggered by infections. Diagnosis is made by the detection of a non-malignant mixed lymphohistiocytic proliferation in the reticuloendothelial system, with evidence of hemophagocytosis. Without treatment, FLH is usually rapidly fatal. The immediate goal of treatment is suppression of the increased inflammatory response by immunosuppressive/immunomodulatory agents and cytotoxic drugs. Genetic cases can only be cured with stem cell transplantation.45 Congenital/infantile fibrosarcoma is a rare tumor that occurs most frequently on the extremities. Lesions are seen less commonly on the head, neck, and trunk, and may also occur in the retroperitoneum.46 The tumor may be present at birth, or may develop during early infancy. MRI is useful in defining the extent of the lesion, but the diagnosis is largely based on histology. Histologic examination reveals a highly cellular fibroblastic proliferation, with sizeable vascular clefts and occasional myxoid degeneration and hemorrhagic necrosis.48 Analysis of the biopsy sample for the recurrent translocation t(12;15)(p13;q25) is highly recommended.49 Clinically, infantile fibrosarcomas are easily confused with either hemangiomas or vascular/lymphatic malformations.50,51 Transient consumptive coagulopathy mimicking Kasabach–Merritt phenomenon has been reported, causing confusion with other vascular tumors (see Chapters 21 and 22).52 The differential diagnosis also includes rhabdomyosarcoma and infantile myofibromatosis. The risk of metastasis, especially in cutaneous lesions, is considerably lower in infants (approx. 8%) than in older patients.53 Treatment consists of wide local excision. Chemotherapy has been used in some patients to reduce the lesion mass preoperatively so as to avoid the need for mutilating surgery, and for lesions that are not resectable.54 Close follow-up to monitor for the presence of local recurrence and metastatic disease, is mandatory.55 Dermatofibrosarcoma protuberans (DFSP) is a fibrohistiocytic tumor with low metastatic potential and a high incidence of local recurrence. Congenital DFSP is rare, but a number of cases have been reported.56–58 Another neoplastic disorder, giant cell fibroblastoma, can also present in early infancy (Fig. 28.7). Some authors consider this to be a variant of DFSP; others consider it to be a separate entity, but believe that hybrids of giant cell fibroblastoma and DFSP may sometimes occur. Characteristically, DFSP begins as an atrophic plaque surrounded by an area of bluish discoloration. Over time, as the cellular proliferation extends into the deep dermis and subcutaneous tissues, nodules develop on the plaque surface (Fig. 28.8). Similar to the lesions seen in older patients, congenital DFSP occurs most commonly on the trunk and proximal extremities.59 Giant cell fibroblastoma more typically presents during infancy as a slow-growing soft-tissue mass. Histologically, DFSP is characterized by the presence of spindle cells in a well-defined and uniform storiform pattern. Focal myxoid change and scarring may occur. Tumor cells express vimentin, but are negative for S100 protein, epithelial membrane antigen, and smooth muscle actin.60 Diagnosis can be facilitated by detection of the collagen type Iα1/platelet-derived growth factor β-chain fusion gene by means of reverse transcriptase–polymerase chain reaction or fluorescence in situ hybridization.58 Clinically, the differential diagnosis of congenital DFSP includes fibrous hamartoma of infancy, infantile myofibromatosis, lipoblastoma (see Chapter 27), and vascular tumors and malformations. Neurofibroma and fibrous histiocytoma may mimic DFSP histologically. Because of the high risk of recurrence, adequate surgical margins must be obtained. Mohs micrographic surgery is the preferred approach in older children. However, in young infants, this can be difficult to arrange logistically in a child requiring general anesthesia for the excision, so wide excision is generally performed.61 Neuroblastoma is among the most common solid tumors of early childhood, and may present at birth or in early infancy.62 In stage 4, neuroblastoma cutaneous metastases present as bluish, firm papules and nodules on the trunk and extremities (Fig. 28.9). Several authors have reported that these lesions may blanch after palpation, and that the blanching persists for 30–60 min.63,64 This phenomenon has been related to the localized release of catecholamines. Periorbital ecchymoses, secondary to orbital metastases, may also occur. Metastatic disease, characterized by fever, hepatomegaly, and failure to thrive, is often present at the time of diagnosis. The primary lesion is usually located in the upper abdomen, arising within the adrenal gland, and may be detected as an enlarging mass. Diagnosis of the cutaneous lesions is based on histologic examination. The dermal or subcutaneous infiltrate consists of small cells with scanty cytoplasm and heterochromatic nuclei. Pseudorosettes and mature ganglion cells are present in differentiated lesions. Immunoperoxidase staining may establish the presence of neuron-specific proteins. Although most cases of neuroblastoma are sporadic, autosomal dominant inheritance may occur. Concordance in monozygotic twins has also been reported.65 The location and extent of the primary lesion is most often established by computed tomography (CT) or MRI. Increased urinary catecholamines are present in the majority of patients. Clinically and histologically, the lesions of neuroblastoma must be differentiated from leukemia and lymphoma. In addition, the blueberry muffin appearance of some lesions may mimic congenital rubella or cytomegalovirus infection. The prognosis of neuroblastoma depends on the age of the patient and the extent of the disease (stages 1–4S). The survival rate at 2 years in children who are diagnosed under the age of 1 year exceeds 80%. In some patients, especially with stage 4S, spontaneous differentiation to neural ganglion cells and regression without treatment have been reported. The choice of treatment depends on staging and patient age, and consists of various combinations of surgery, radiation therapy, and chemotherapy.66 Rhabdomyosarcoma (RMS) presents most commonly as a tumor of the head and neck (Fig. 28.10A,B) There are two histologic subtypes: embryonal RMS, which is more common and usually has a better prognosis, and alveolar RMS, which is characterized by the t(2;13)(q35;q14) and t(1;13)(p36;q14) chromosomal translocations.67 An association between RMS and both neurofibromatosis type 1 and major congenital abnormalities has been observed.68 RMS arising in children with giant congenital melanocytic nevi has also been reported.69 A cutaneous origin of RMS is rare. Most often, extension of the tumor into the dermis results in the evolution of a nodule or plaque.70 Facial lesions are most common and these must be differentiated clinically from dermoid cysts, hemangiomas, and inflammatory disorders. Histologically, there is a dermal infiltrate of small blue cells with occasional differentiation toward rhabdomyoblasts. The presence of staining for desmin, vimentin and muscle actin help to differentiate these lesions from neuroblastoma and lymphoma.71 Treatment is based on the extent of local, regional, and distant disease, and consists of a combination of surgery, radiation, and chemotherapy. Combined modality therapy results in a long-term survival of >50%.72 Rhabdoid tumor is a rare and highly malignant neoplasm with rapid growth and early metastases. The skin may be the site of a primary tumor or of metastases from other locations (Fig. 28.10C). The majority of neonates are found to have metastatic disease at the time of diagnosis, and brain tumors are a frequent finding. Characteristic bluish cutaneous nodules may be located over the entire skin surface. The tissue of rhabdoid tumors has a characteristic genetic alteration in the region 11.2 of the long arm of chromosome 22 (22q11.2), characterized by the deletion or mutation of the hSNF5/SMARCB1/INI1 gene resulting in the loss or reduced expression of INI protein. The survival rate is extremely low.49,71 Malignant melanoma is very rare in neonates and infants but can develop in several different clinical situations73–75: 1. Congenital malignant melanoma arising de novo. Malignant melanoma may present at birth as a nodular, darkly pigmented, rapidly growing skin lesion sometimes with ulceration.76,77 In these neonates, there is no clinical or histologic evidence of an underlying congenital melanocytic nevus, and there is no maternal history of melanoma (Fig. 28.11). 2. Congenital malignant melanoma arising in a giant congenital melanocytic nevus. In these patients, ulcerated and nonulcerated nodules may be present in the congenital melanocytic nevus and on adjoining skin. The presence of prenatal metastatic disease has been noted to occur in some children.78,79 4. Congenital malignant melanoma secondary to maternal melanoma. Transplacental transmission from a mother with metastatic disease may occur.80 Typically, the skin lesions are multiple pigmented macules, papules, and nodules, and there may be multiorgan involvement. 5. Spitzoid melanoma is a relatively rare subtype which shares histopathologic features with Spitz nevus.81 The prognosis is variable; Spitzoid melanoma with regional lymph node involvement and no further progression has been reported.82 However, some cases have resulted in a fatal outcome. The diagnosis of malignant melanoma is made by excisional biopsy of the suspicious lesion, and the cellular and architectural features are similar to those seen in melanoma in older patients. However, a wide variety of benign and malignant tumors, with small round cell, spindled, neural, and epithelioid components, have been observed within congenital melanocytic nevi.77 In addition, histologic changes within a benign, congenital melanocytic nevus may include displaced large melanocytes within the epidermis, and heterogeneous patterns of melanocytic hyperplasia.78 In some cases, findings of this type have led to an incorrect diagnosis of malignant melanoma. Biopsies of congenital melanocytic nevi, especially in the neonate, must therefore be interpreted with caution.73,83,84 The evaluation of children with all forms of melanoma must include a complete evaluation for local and distant lymph node involvement. Metastases are most frequently seen in the CNS, bones, lungs, and liver. Treatment is based on lesion thickness and the stage of the disease. Therapeutic options include lymph node dissection of enlarged draining regional nodes and chemotherapy for metastatic disease.79 Lesions arising de novo have an unpredictable prognosis, and long-term survival has been reported in children who developed both local recurrences and metastatic lesions. Melanomas arising in congenital melanocytic nevi appear to have a significantly worse prognosis. Congenital malignant melanoma secondary to maternal melanoma is usually fatal, but spontaneous regression has been reported to occur.80 Congenital teratomas most frequently present as masses in the cervical, nasopharynx, or sacrococcygeal region.85 These are usually benign tumors, which are derived from elements of all three germinal layers, and which result in significant morbidity and mortality because of their location, size, and tendency to cause airway obstruction. Approximately 10–20% of SCT may contain primitive germ cell tumor components, and can evolve to actual malignancy. This risk increases significantly with age at diagnosis. The diagnosis requires complete pathologic examination of the entire resected tumor for malignant change. The differential diagnosis includes dermoids and lymphatic or vascular birthmarks. Treatment consists of complete surgical excision.86 Mastocytosis refers to a disease spectrum with varied presentations, all characterized by infiltration of benign mast cells (MC) in the skin or other organs. ‘Mastocytosis in the skin’ or MIS is the designation given to patients who have cutaneous MC lesions and have not been evaluated for systemic involvement (e.g., bone marrow examination), typically the case in children. A total of 55% of cases develop during the first 2 years, and an additional 10% develop before puberty.87 Cases developing after puberty are classified as adult onset. Both sexes are affected equally. MIS is subclassified into maculopapular cutaneous mastocytosis (MPCM, also known as urticaria pigmentosa, UP), solitary mastocytoma, and diffuse cutaneous mastocytosis (DCM). However, many display overlapping features. MPCM is the most common clinical manifestation of mastocytosis. Solitary mastocytoma is less common, although it may be underreported, and DCM, the most severe form, is rare. Most cases are sporadic; however, there are several reports of MPCM affecting multiple family members, and some believe DCM can be inherited in an autosomal dominant fashion.87–89 Telangiectasia macularis eruptiva perstans (TMEP) has been reported rarely in the neonatal period, with onset typically during adulthood. Most patients with MIS exhibit Darier’s sign, which is the development of an urticarial wheal and flare after firm stroking of lesional skin. This cutaneous finding represents the response to physical disruption of the granular contents of mast cells, particularly histamine. Rarely, flushing and hypotension have resulted from stroking of a large lesion or from surgery. Some patients display dermatographism, the formation of linear urticarial plaques following scratching of uninvolved skin. However, this nonspecific finding also occurs in up to 5% of the general population. A variety of physical stimulants and drugs can evoke mast cell degranulation, resulting in urtication, bulla formation, or systemic manifestations (flushing, hypotension, or shock) (Box 28.1). Box 28.1 Histamine-releasing triggers to avoid in mastocytosis Physical stimuli
Neoplastic and Infiltrative Diseases
Introduction
Neoplastic disorders
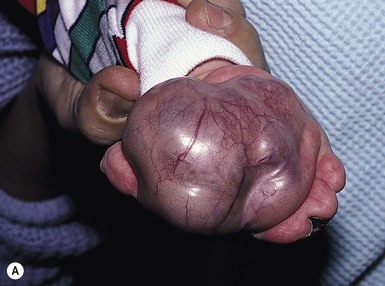
Leukemia
Cutaneous findings
Diagnosis
Treatment and course
Leukemia in infancy
Langerhans’ cell histiocytosis
Cutaneous findings
Congenital ‘self-healing’ langerhans’ cell histiocytosis
Extracutaneous findings
Diagnosis
Differential diagnosis
Treatment and course
Lymphoma
Mycosis fungoides
Primary cutaneous anaplastic large cell lymphoma
Lymphomatoid papulosis
Cutaneous B-cell lymphoma
Hemophagocytic lymphohistiocytosis
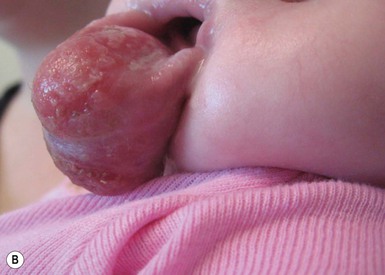
Infantile fibrosarcoma
Diagnosis
Differential diagnosis
Treatment and course
Dermatofibrosarcoma protuberans
Cutaneous findings
Diagnosis
Differential diagnosis
Treatment
Neuroblastoma
Cutaneous findings
Extracutaneous findings
Diagnosis
Differential diagnosis
Treatment and course
Rhabdomyosarcoma
Rhabdoid tumor
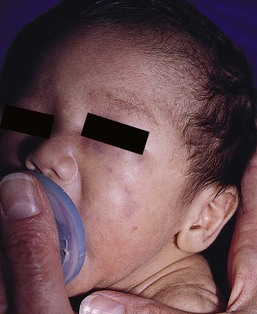
Malignant melanoma
Congenital teratoma
Infiltrative disorders
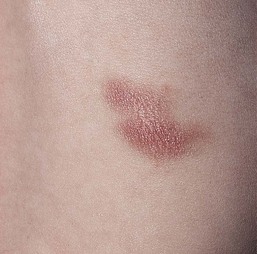
Mastocytosis
Cutaneous findings
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree