Somatic mutations in genes of the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway cause segmental overgrowth, hamartomas, and malignant tumors. Mosaicism for activating mutations in AKT1 or PIK3CA cause Proteus syndrome and PIK3CA-Related Overgrowth Spectrum, respectively. Postzygotic mutations in PTEN or TSC1/TSC2 cause mosaic forms of PTEN hamartoma tumor syndrome or tuberous sclerosis complex, respectively. Distinct features observed in these mosaic conditions in part reflect differences in embryological timing or tissue type harboring the mutant cells. Deep sequencing of affected tissue is useful for diagnosis. Drugs targeting mTORC1 or other points along this signaling pathway are in clinical trials to treat these disorders.
Key points
- •
Mosaicism may be considered in sporadic cases; there should be a negative history in ancestral generations or siblings but offspring may be affected through gonadal mosaicism.
- •
A patchy distribution of cutaneous features and tissue overgrowth or disseminated disease that is mild should raise suspicion for mosaicism.
- •
Next-generation sequencing, or other methods for detecting low frequency alleles, of affected tissue is frequently necessary to identify the genetic basis of mosaic conditions.
- •
Drugs that target proteins along the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway have shown promise in the treatment of these disorders.
| BRRS | Bannayan-Riley-Ruvalcaba Syndrome |
| KTS | Klippel-Trenaunay syndrome |
| mTORC1 | Mechanistic target of rapamycin complex 1 |
| PHTS | PTEN hamartoma tumor syndrome |
| PI3K | Phosphoinositide-3-kinase |
| PIP2 | Phosphatidylinositol (4,5)-bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PROS | PIK3CA-related overgrowth spectrum |
| Rheb | Ras homolog enriched in brain |
| TSC | Tuberous sclerosis complex |
Introduction
Mosaicism may occur as a somatic mutation occurring during embryogenesis, resulting in an organism composed of 2 (or more) genetically distinct cell lineages. The resulting phenotype depends on the numbers and organization of abnormal cells in relation to normal cells and how the mutation affects cellular function. When the mutation affects cell signaling pathways regulating cell growth, apoptosis, or migration, dramatic regional alterations in the appearance of the skin can occur sometimes with regional overgrowth or tumor susceptibility. Dermatologists are frequent observers of these mosaic conditions and play important roles in diagnosis and management.
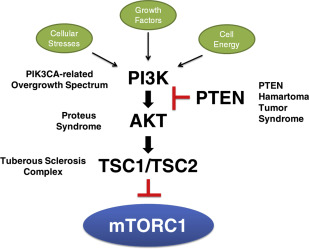
Somatic mutations in any of the genes in the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway ( Fig. 1 ) may result in a spectrum of abnormal growth, ranging from an isolated small skin lesion with minimal or no overgrowth to extensive skin involvement with striking extremity enlargement and tumor susceptibility. Proteus syndrome, caused by mutations in AKT1 , and the PIK3CA -related overgrowth spectrum (PROS) may be considered archetypal mosaic disorders, given the patchy distribution of disease features. Tuberous sclerosis complex (TSC), caused by mutations in either TSC1 or TSC2 , and PTEN hamartoma tumor syndrome (PHTS) are most frequently associated with germline mutations, but mosaic forms also appear as isolated (simplex or sporadic) occurrences. This article primarily focuses on these entities, chosen for their shared abnormalities in a signaling pathway and analogous or overlapping phenotypes.
Introduction
Mosaicism may occur as a somatic mutation occurring during embryogenesis, resulting in an organism composed of 2 (or more) genetically distinct cell lineages. The resulting phenotype depends on the numbers and organization of abnormal cells in relation to normal cells and how the mutation affects cellular function. When the mutation affects cell signaling pathways regulating cell growth, apoptosis, or migration, dramatic regional alterations in the appearance of the skin can occur sometimes with regional overgrowth or tumor susceptibility. Dermatologists are frequent observers of these mosaic conditions and play important roles in diagnosis and management.
Somatic mutations in any of the genes in the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway ( Fig. 1 ) may result in a spectrum of abnormal growth, ranging from an isolated small skin lesion with minimal or no overgrowth to extensive skin involvement with striking extremity enlargement and tumor susceptibility. Proteus syndrome, caused by mutations in AKT1 , and the PIK3CA -related overgrowth spectrum (PROS) may be considered archetypal mosaic disorders, given the patchy distribution of disease features. Tuberous sclerosis complex (TSC), caused by mutations in either TSC1 or TSC2 , and PTEN hamartoma tumor syndrome (PHTS) are most frequently associated with germline mutations, but mosaic forms also appear as isolated (simplex or sporadic) occurrences. This article primarily focuses on these entities, chosen for their shared abnormalities in a signaling pathway and analogous or overlapping phenotypes.
PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway and overgrowth
Cell growth is mediated by extracellular cues and may occur via increasing cell mass or cell division or by suppression of apoptosis. Mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) is a central regulator of cell growth that is disrupted in many human disorders of cell proliferation, including cancers. Under normal conditions, mTORC1 is sensitive to inputs from diverse cellular and environmental cues (see Fig. 1 ), including the phosphoinositide-3-kinase (PI3K) pathway. In short, growth factors stimulate PI3K, which then converts phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) and permits activation of AKT. PTEN dephosphorylates PIP3 to PIP2, thereby exerting inhibitory control on AKT. AKT phosphorylates several proteins, including the TSC1-TSC2 complex, and thus alleviates negative control on mTOR to promote cell growth (see Fig. 1 ).
PTEN , TSC1 , and TSC2 are tumor suppressors. For each of these genes, a germline mutation causes a loss of function allele, leading to a syndrome with susceptibility to multiple tumors. Tumorigenesis involves inactivation of the second allele, typically via a second-hit somatic mutation in the wild-type allele. Loss of function of PTEN results in increased levels of PIP3 and alleviates inhibitory control on AKT to cause tumor formation in PHTS. Biallelic mutations in TSC1 or TSC2 result in increased Ras homolog enriched in brain (Rheb)-GTP, which activates signaling through mTORC1, causing tumor formation in TSC. Although the PHTS and TSC are typically caused by germline mutations, both can have mosaic presentations, often with mild disease or later onset than inherited disease.
PIK3CA , a gene encoding the catalytic subunit of PI3K, and AKT1 are oncogenes and thus a gain of function, or activating mutation in only one copy of the allele is the mechanism of disease. Strongly activating mutations in these genes may only be seen in patients through mosaicism. Point mutations activating PIK3CA have been documented in a variety of mosaic disorders captured under the umbrella term PROS. The mosaic presence of an activating mutation in AKT1 results in Proteus syndrome.
Phenotype of disorders of the PI3K/PTEN/AKT/TSC/mTORC1 signaling pathway
Mosaic-Only Oncogenes
It is widely regarded that strongly activating germline mutations in AKT1 and PIK3CA are lethal, with few reported exceptions. Mutations in these genes result in distinctive segmental, or asymmetric overgrowth syndromes that can involve the bone, muscle, adipose tissue, skin, and/or nerves and may be relatively stable ( PIK3CA ) or progressive ( AKT1 or PIK3CA ) in course. The severity of the overgrowth may range from a slightly enlarged digit to a gigantic limb. Hamartomas, or benign, focal overgrowths resembling their tissue of origin, may also be present. The epidermal nevus and vascular malformation in these disorders ( Fig. 2 A, B ) are examples of the Blaschkoid and checkerboard patterns of cutaneous mosaicism, respectively.
Somatic activation in AKT1 has been linked to Proteus syndrome, an extraordinarily rare disorder (incidence is <1 case per 10 million) that is characterized by sporadic occurrence, asymmetric distribution of lesions, and progressive course. Frequent dermatologic lesions include cerebriform connective tissue nevi, lipomas, linear keratinocytic epidermal nevus, and lymphovascular malformations.
PROS represents a broad constellation of somatic disorders caused by activating mutations in PIK3CA , including congenital lipomatous overgrowth, vascular malformations, linear keratinocytic epidermal nevi, and skeletal or spinal anomalies (CLOVES) syndrome; fibroadipose hyperplasia or overgrowth; hemihyperplasia multiple lipomatosis; certain megalencephaly syndromes; isolated macrodactyly; isolated lymphatic malformations; seborrheic keratoses; and benign lichenoid keratoses. The phenotype of this spectrum ranges from isolated disease, such as macrodactyly, megalencephaly, or vascular malformations, to syndromes defined by tissue overgrowth, vascular malformations, and epidermal nevi. Because several individuals with Klippel-Trenaunay syndrome (KTS), which bears clinical semblance to PROS, have pathogenic PIK3CA mutations, KTS may be under the PROS umbrella.
Germline or Mosaic Tumor Suppressor Genes
PHTS represents a spectrum of diseases caused by mutations in the PTEN tumor suppressor gene, including Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome (BRRS). Cowden syndrome is an autosomal-dominant condition characterized by skin hamartomas and mixed benign and malignant tumors of the thyroid, breast, and endometrium. Characteristic mucocutaneous lesions include trichilemmomas, acral keratoses, and oral papillomatosis, and are usually present by the third decade. Malignancy risk is greatest for breast cancer (approximately 85%) and is increased for thyroid cancer, endometrial cancer, colon cancer, and melanoma.
BRRS is usually congenital in onset and cutaneous manifestations include pigmented macules of the glans penis, although formal diagnostic criteria have not yet been defined. Given the clinical overlap and known allelism of BRRS and Cowden syndrome, some assert that they are 1 condition with distinct manifestations in childhood and adulthood, respectively.
For conditions with autosomal-dominant inheritance, 3 categories of mosaicism exist ( Fig. 3 ). Disseminated mosaicism is perhaps the most common; the phenotype is usually clinically similar to germline disease with multiple lesions and/or tumors. Segmental mosaicism may be categorized based on the genetic background of the organism. Type 1 segmental mosaicism describes a localized postzygotic heterozygous mutation in an organism with 2 otherwise normal alleles, resulting in expected disease manifestations restricted to a discrete region. Type 2 segmental mosaicism describes an organism with a localized postzygotic mutation in trans (ie, on the otherwise normal allele) to a mutant allele that was inherited from a heterozygous parent, resulting in biallelic mutations in some cell lines. If the heterozygous mutant state causes disease, as in TSC or PHTS, then type 2 segmental mosaicism results in a clinical phenotype with a more severe segment of disease in a patient with otherwise typical disease distribution.
Mosaicism has been genetically confirmed in several cases among the 10% to 40% of patients with de novo mutations in PTEN . Most reports describe disseminated mosaicism, resulting in an anatomically diffuse distribution of cutaneous hamartomas and mixed internal lesions that are clinically difficult to distinguish from inherited or de novo, nonmosaic Cowden syndrome disease. Interestingly, there have also been isolated occurrences of patients with germline PTEN mutations that harbor loss of PTEN heterozygosity in lesions not characteristic of Cowden syndrome or BRRS. Because of its distinct clinical picture, type 2 segmental mosaicism for PTEN was termed segmental overgrowth, lipomatosis, arteriovenous malformations, and epidermal nevi (SOLAMEN) syndrome to reflect the presence of these features in addition to those consistent with a germline PTEN mutation. Similarly, an overgrowth of fat, blood vessels, and fibrous tissue (termed the PTEN hamartoma of soft tissue) has been described in individuals with Cowden syndrome and BRRS, although type 2 segmental mosaicism of the PTEN gene has not been molecularly confirmed in these lesions.
TSC is a neurocutaneous syndrome inherited in an autosomal-dominant pattern characterized by hamartomas in multiple organ systems, including the brain, kidneys, lungs, and skin. Hypomelanotic macules, angiofibromas, shagreen patches, ungual fibromas, and fibrous cephalic plaques are among the most frequent and specific cutaneous findings. Oral findings include oral fibromas and dental enamel pits. Noninvasive, visceral hamartomas can be harmful due to hemorrhage risk or impingement on adjacent structures. A subset of TSC patients have no mutation identified using conventional analysis and many of these patients are mosaic. A postzygotic mutation in TSC1 or TSC2 before neural crest cell differentiation may explain disseminated TSC-related skin lesions, including bilateral angiofibromas ( Fig. 2 C), ungual fibromas, and hypomelanotic macules. Unilateral facial angiofibromas are suspected to represent type 1 segmental mosaicism ( Fig. 2 D, E).
Comparing phenotypes
Although these disorders are distinctive, there is overlap in the phenotypes associated with mutations of PIK3CA , PTEN , TSC1 , TSC2 , and AKT1 , which may be expected given their common pathway and mosaic pathogenesis. Disorders of this axis may be broadly characterized by frequency of 1 or more of the following features: segmental overgrowth, hamartomas, or malignant tumors ( Table 1 ). Because all 4 conditions are characterized by hamartomas, Table 2 contrasts the specific types of hamartomas observed in these conditions.
| PIK3CA | PTEN | AKT1 | TSC1/TSC2 | |
|---|---|---|---|---|
| PROS | PHTS | Proteus Syndrome | Tuberous Sclerosis Complex | |
| Segmental Overgrowth | *** | * | *** | * |
| Hamartomas | *** | *** | *** | *** |
| Malignant Tumors | * | *** | * | * |
Related posts:
 Basic Science Insights into Clinical Puzzles
Basic Science Insights into Clinical Puzzles
 Establishing Tolerance to Commensal Skin Bacteria
Establishing Tolerance to Commensal Skin Bacteria
 Dermatologic Manifestations of Monogenic Autoinflammatory Diseases
Understanding Inherited Cylindromas
Interleukin-22 and Cyclosporine in Aggressive Cutaneous Squamous Cell Carcinoma
Dermatologic Manifestations of Monogenic Autoinflammatory Diseases
Understanding Inherited Cylindromas
Interleukin-22 and Cyclosporine in Aggressive Cutaneous Squamous Cell Carcinoma
 Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence
Melanocytic Nevi and the Genetic and Epigenetic Control of Oncogene-Induced Senescence
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


