Molecular Pathology of the Breast
Mohiedean Ghofrani
Although histopathologic examination of breast tumors has been practiced since the 19th century (1), the application of ancillary techniques such as immunohistochemistry (IHC) and more recently molecular diagnostics including gene expression profiling as adjuncts to surgical pathology have greatly expanded our understanding of breast cancer (2). Conventional microscopy methods to evaluate morphologic characteristics such as histologic subtype, architectural differentiation, cytologic atypia, and mitotic activity in routine hematoxylin and eosin (H&E)-stained glass slides had already demonstrated the wide variety of histologic presentations of breast cancer, suggesting that breast cancer is not one disease but rather a multitude of diseases that happen to arise in the breast, each with a different prognosis and response to therapy (3).
This concept was further reinforced with the advent of IHC in the latter part of the 20th century, a diagnostic technique that highlights the expression of specific antigens that are not readily detected by conventional staining methods such as H&E. IHC not only provided prognostic information (for example, that estrogen receptor [ER]-negative or human epidermal growth factor receptor 2 [HER-2]–positive tumors have a poorer prognosis) but also predictive information (e.g., that ER-positive or HER-2–positive tumors are more likely to respond to tamoxifen or trastuzumab, respectively). The College of American Pathologists (CAP) and American Society of Clinical Oncology (ASCO) subsequently developed guidelines to standardize testing and reporting of ER, progesterone receptor (PR) and HER-2, which are updated regularly (4,5).
At the beginning of the 21st century, the tools of gene expression profiling were applied to a variety of breast tumors leading to the theory that different types of breast cancer can be prognostically categorized into certain “intrinsic” molecular subtypes. Based on these and other discoveries, several commercial tests have been developed to help elucidate the molecular characteristics of each individual patient’s tumor, specifically its risk of recurrence, mortality, and by extension, benefit from cytotoxic chemotherapy, thereby paving the way to realize the promise of “personalized medicine.”
Molecular Diagnostics
From a certain viewpoint, techniques such as IHC, which highlight the presence of specific protein molecules, may be considered “molecular” diagnostic methods. By this logic, it may even be argued that the utility of traditional H&E and other histochemical stains depends on distinctive molecular features. However, for the purposes of this chapter, molecular pathology will be defined as the study of disease through the examination of nucleic acids, namely deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), in their various forms.
One of the tenets of molecular oncology is that cancer is the result of nonlethal mutations in DNA that amplify the expression of oncogenes or inactivate tumor suppressor genes, both of which lead to uninhibited cellular proliferation. A variety of basic molecular techniques have been employed over the past few decades for the study of cancer, first in the research laboratory, and now incorporated in commercially available clinical tests. These include various methods for nucleic acid extraction, separation (i.e., resolution), detection, characterization, amplification, and sequencing. Of the variety of existing molecular techniques, this chapter will first briefly describe two methods that are most commonly being utilized in breast cancer molecular testing—the polymerase chain reaction (PCR) and gene expression profiling—and then present an overview of the more common clinically available molecular tests for breast cancer.
Polymerase Chain Reaction
During normal DNA replication, the hydrogen bonds that hold two strands of DNA in the double helix together are split, exposing the two strands to serve as templates for addition of new complementary nucleotides by the enzyme DNA polymerase, ultimately resulting in the formation of two newly replicated strands. Since each new double helix that is formed is composed of one parent strand and one newly replicated strand, this replicative process is semiconservative, which to a large extent
maintains the sequence of nucleotides in DNA through successive generations.
maintains the sequence of nucleotides in DNA through successive generations.
The PCR takes advantage of this natural process to replicate, that is, amplify segments of nucleic acid in vitro (6). A typical PCR goes through multiple cycles, whereby the amount of target DNA is doubled in each cycle. In the first step of a typical cycle, target DNA is exposed to a temperature of around 95°C, which leads to denaturation of the double helix, that is, the hydrogen bonds between the two parent strands are broken (Fig. 3-1a). In step 2, the temperature is reduced to 50° to 70°C, so two primers, which are specific short sequences of DNA that determine the start point and end points of the amplification process, form hydrogen bonds (“anneal”) with their specific complementary sequence in the DNA template strands (Fig. 3-1b). In the third step, temperatures are increased to 68° to 72°C, the optimal temperature for the polymerase enzyme to extend a new DNA strand from the primers (Fig. 3-1c). Then, the cycle is repeated. Given that the duration of each step of the cycle—denaturation, annealing, and extension—is measured in seconds, and that the amount of target doubles in each successive cycle, automated PCR instruments (thermal cyclers) can now produce millions of copies, or amplicons, of a specific target sequence within 1 or 2 hours (7).
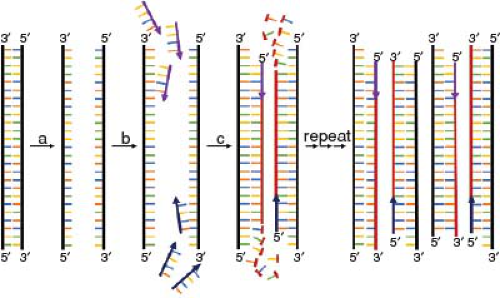 FIGURE 3-1 The polymerase chain reaction (PCR) is an efficient molecular technique to amplify (i.e., replicate) a specific DNA sequence in the genome. In each PCR cycle DNA is first “denatured” (a), whereby the two strands of DNA are separated through exposure to increased temperature. Then, the temperature is lowered so two specific primers (purple and blue arrows) “anneal” (b) to their complementary sequences in the two DNA templates (black). Finally, the enzyme DNA polymerase (not pictured) facilitates the building or “extension” (c) of new DNA strands (red) by attaching complementary nucleotides starting from the primers and continuing in the 5′ to 3′ direction, leading to two double strands of DNA that contain the sequence delimited by the two primers. Each time the cycle is repeated, the DNA strands formed in the previous cycles, in turn, serve as templates for the creation of new DNA strands. |
Several variants of PCR have been developed. These include:
Multiplex PCR, in which more than one primer pair are added to the reaction environment so that multiple target sequences are amplified simultaneously;
Reverse transcriptase (RT-)PCR, where the initial target is RNA instead of DNA, and the enzyme RT is used to essentially perform the mirror opposite of normal transcription, that is, to form complementary DNA (cDNA) off of an RNA template rather than messenger RNA (mRNA) off of a DNA template;
Nested PCR, in which two pairs of primers are used in two successive rounds of amplification—the second pair targeting sequences located slightly inside the first primer pair to increase sensitivity and specificity of the reaction; and
Real-time or quantitative PCR (which is confusingly also abbreviated as RT-PCR, but more appropriately as qPCR or RT-qPCR) where the amount of amplicon is monitored in real time using a fluorescent marker, so that based on the cycle number in which fluorescence exceeds a certain threshold, the starting amount of target in an unknown specimen can be calculated—the assumption being that the more target in the initial sample, the fewer cycles that would have been necessary for the fluorescence to exceed the threshold (Fig. 3-2).
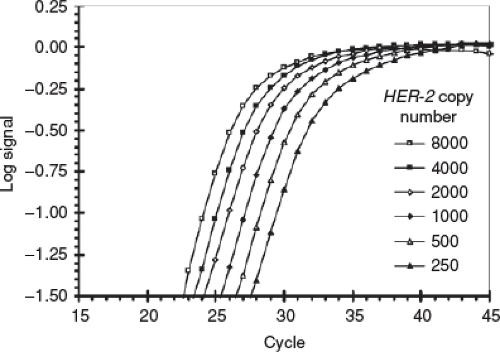 FIGURE 3-2 Real time quantitative PCR (RT-qPCR) is a type of PCR in which the quantity of a specific nucleic acid sequence in a test sample can be estimated. At the end of each PCR cycle, the amount of amplicon is measured, and based on the cycle number in which that quantity exceeds a certain threshold, the initial quantity can be calculated, since the higher the amount of targeted nucleic acid in the initial sample, the earlier the measured amplicon will exceed the quantitative threshold. (From Königshoff M, Wilhelm J, Bohle RM, et al. HER-2/neu gene copy number quantified by real-time PCR: comparison of gene amplification, heterozygosity, and immunohistochemical status in breast cancer tissue. Clin Chem 2003;49:219–229.) |
Gene Expression Profiling
Gene expression profiling is a method whereby the expression of thousands of genes can be simultaneously measured in a patient’s tissue sample. This is achieved using a DNA microarray or “chip,” a collection of microscopic DNA spots attached to a solid surface typically no larger than the size of a postage stamp (Fig. 3-3). Each of these DNA spots contains a very small amount (on the order of 10−12 moles, a picomole) of a specific DNA sequence or “probe.” DNA or RNA from a tissue sample is applied to this DNA microarray under controlled conditions so that complementary sequences in the sample hybridize (i.e., form hydrogen bonds) with their respective probe in the microarray.
The amount of hybridization in each spot can be quantified and then visualized in a heat map, in which, for example, overexpression of a certain gene is shown as a red dot while underexpression is shown as a green dot. Since the gene expression profile of a tumor sample will be different from normal tissue, each tumor’s aggregate pattern of gene overexpression or underexpression will form that tumor’s unique gene expression profile. By extension, the gene expression profile of hundreds of different tumor and normal tissue samples can be compared side by side in a two-dimensional heat map, in which each column represents a sample and each row represents a gene. Complex bioinformatic algorithms can identify a subset of genes that are differentially expressed in tumors and sort the different tumor samples in the heat map based on their similarity in expressing these informative genes so that a hierarchical clustering emerges (Fig. 3-4). By correlating the shared gene expression profile of these clusters of tumor samples with clinical outcomes, it can be shown that each molecular subtype is typically associated with a characteristic natural history, metastatic pattern, and sensitivity to different treatments, demonstrating the causal relationship between genotype and phenotype.
Related posts:
 Axillary Nodal Recurrence
Axillary Nodal Recurrence
 The Surgical Management of Locally Advanced and Stage IV Breast Cancer
The Surgical Management of Locally Advanced and Stage IV Breast Cancer
 Alternative Methods for Axillary Sentinel Lymph Node Dissection
Alternative Methods for Axillary Sentinel Lymph Node Dissection
 Optimizing Mastectomy Flaps Based on Breast Anatomy
Optimizing Mastectomy Flaps Based on Breast Anatomy
 Liposuction as a Viable Treatment for End-Stage Upper Extremity Lymphedema
Liposuction as a Viable Treatment for End-Stage Upper Extremity Lymphedema
 Oncoplastic Techniques in Breast Conservation Therapy—The Plastic Surgeon’s Perspective
Oncoplastic Techniques in Breast Conservation Therapy—The Plastic Surgeon’s Perspective
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


